Cellular and Molecular Gastroenterology and Hepatology ( IF 7.1 ) Pub Date : 2020-05-22 , DOI: 10.1016/j.jcmgh.2020.05.005 Usman Yaqoob 1 , Fanghong Luo 2 , Thomas Greuter 3 , Nidhi Jalan Sakrikar 1 , Tejasav S Sehrawat 1 , Jianwen Lu 1 , Xiao Hu 1 , Jinhang Gao 1 , Enis Kostallari 1 , Jingbiao Chen 1 , Juan Pablo Arab 1 , Rosa Martin-Mateos 4 , Sheng Cao 1 , Vijay H Shah 1
|
Background & Aims
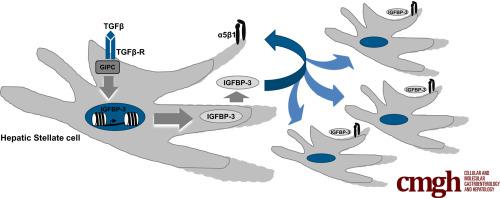
Transforming growth factor (TGF-β)–induced activation of quiescent hepatic stellate cells (HSCs) and their transformation to myofibroblasts is a key event in liver fibrosis and portal hypertension. GIPC (also referred to as synectin) is a downstream signal activation molecule of TGF-β and other receptors. In this study, we sought to identify novel genes targeted by TGF-β and GIPC and elucidate if and how they may contribute to liver fibrosis.
Methods
We performed sequential messenger RNA sequencing analysis on TGF-β–stimulated HSCs and then on TGF-β–stimulated HSCs in the presence and absence of GIPC also referred to as synectin (GIPC) knockdown. Insulin-like growth factor binding protein-3 (IGFBP-3) transport protein emerged as a top activation target of both TGF-β and GIPC. Quantitative polymerase chain reaction, enzyme-linked immunosorbent assay, targeted chromatin immunoprecipitation, and Western blot analysis were done for further confirmation.
Results
IGFBP-3, an insulin growth factor transport protein, emerged as a top activation target of both TGF-β and GIPC, which was confirmed by quantitative polymerase chain reaction, enzyme-linked immunosorbent assay, and Western blot analysis. Targeted chromatin immunoprecipitation showed that GIPC increases the histone 3 lysine 27 (H3K27) acetylation activating mark and concurrently decreases the H3K27 inhibitory trimethylation (H3K27m3) mark, providing an epigenetic correlate to the gene regulation changes. In vivo, global knockout of IGFBP-3 mice resulted in attenuation of HSC activation markers and attenuation of portal pressure in response to chronic liver injury models. Analysis of serum levels from cirrhotic patients also showed an IGFBP-3 increase of more than 2-fold compared with healthy controls. Finally, in vitro mechanism studies showed that IGFBP-3 promotes HSC migration through integrin-dependent phosphorylation of protein kinase B.
Conclusions
TGF-β up-regulates IGFBP-3 through GIPC, leading to increased HSC migration in vitro and promotes portal hypertension in vivo. These studies support the role of IGFBP-3 as a potential pathophysiologic target or biomarker in chronic liver disease.
中文翻译:
GIPC 调节的 IGFBP-3 通过 β1-整合素途径促进体外 HSC 迁移和体内门脉高压。
背景与目标
转化生长因子 (TGF-β) 诱导静止肝星状细胞 (HSC) 的激活及其向肌成纤维细胞的转化是肝纤维化和门脉高压的关键事件。 GIPC(也称为 Synectin)是 TGF-β 和其他受体的下游信号激活分子。在这项研究中,我们试图鉴定 TGF-β 和 GIPC 靶向的新基因,并阐明它们是否以及如何促进肝纤维化。
方法
我们对 TGF-β 刺激的 HSC 进行了序贯信使 RNA 测序分析,然后在存在和不存在 GIPC(也称为联连蛋白 (GIPC) 敲低)的情况下对 TGF-β 刺激的 HSC 进行了连续信使 RNA 测序分析。胰岛素样生长因子结合蛋白 3 (IGFBP-3) 转运蛋白成为 TGF-β 和 GIPC 的首要激活靶标。进行定量聚合酶链反应、酶联免疫吸附测定、靶向染色质免疫沉淀和Western blot分析进行进一步证实。
结果
IGFBP-3(一种胰岛素生长因子转运蛋白)成为 TGF-β 和 GIPC 的首要激活靶标,定量聚合酶链反应、酶联免疫吸附测定和蛋白质印迹分析证实了这一点。靶向染色质免疫沉淀显示,GIPC 增加了组蛋白 3 赖氨酸 27 (H3K27) 乙酰化激活标记,同时降低了 H3K27 抑制性三甲基化 (H3K27m3) 标记,提供了与基因调控变化相关的表观遗传。在体内,IGFBP-3 小鼠的整体敲除导致 HSC 激活标记物减弱以及对慢性肝损伤模型的门静脉压力减弱。对肝硬化患者血清水平的分析还显示,与健康对照相比,IGFBP-3 增加了 2 倍以上。最后,体外机制研究表明IGFBP-3通过整合素依赖性蛋白激酶B磷酸化促进HSC迁移。
结论
TGF-β通过GIPC上调IGFBP-3,导致体外HSC迁移增加并促进体内门静脉高压。这些研究支持 IGFBP-3 作为慢性肝病潜在病理生理学靶点或生物标志物的作用。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号