当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Computational studies on the conformational preference of N‐(Thiazol‐2‐yl) benzamide
Journal of the Chinese Chemical Society ( IF 1.6 ) Pub Date : 2020-05-04 , DOI: 10.1002/jccs.201900277 Afsaneh Zonouzi 1, 2 , Ali Kakeshpour 1 , Parviz Rashidi Ranjbar 1 , Ashraf Moradi 3
Journal of the Chinese Chemical Society ( IF 1.6 ) Pub Date : 2020-05-04 , DOI: 10.1002/jccs.201900277 Afsaneh Zonouzi 1, 2 , Ali Kakeshpour 1 , Parviz Rashidi Ranjbar 1 , Ashraf Moradi 3
Affiliation
|
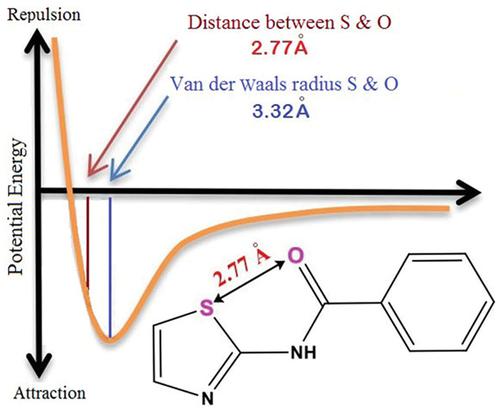
N‐(Thiazol‐2‐yl) benzamide 1 substructures are found in some of bioactive compounds. In some of protein/ligand co‐crystals, the 1 moiety adopts a conformer in which the amide O and the thiazole S atoms are close. In fact, in the crystalline structure of 1, the O—S distance is even shorter than Van der Waals radius. Although the natural bond orbital analysis finds a weak stabilizing interaction between O and S atoms, the attractive dipole–dipole interaction between the amide N─H and thiazole N atom seems to play a more significant role. Moreover, an intramolecular O—H hydrogen bonding in dimeric forms found to have an important role in the conformation preference of 1. Computational details for the stability of conformers have been discussed using quantum theory of atoms in molecules, natural bond orbital (NBO) and noncovalent interaction index analysis.
中文翻译:

N-(噻唑-2-基)苯甲酰胺构象偏好的计算研究
在某些生物活性化合物中发现了N-(噻唑-2-基)苯甲酰胺1亚结构。在某些蛋白质/配体共晶体中,1部分采用的构象异构体中酰胺O和噻唑S原子紧密相连。实际上,在结晶结构1中,O-S距离比范德华半径更短。尽管自然键轨道分析发现O和S原子之间的稳定相互作用较弱,但酰胺N–H和噻唑N原子之间有吸引力的偶极-偶极相互作用似乎起着更重要的作用。此外,发现二聚体形式的分子内OH氢键对1的构象偏好具有重要作用。使用分子中原子的量子理论,自然键轨道(NBO)和非共价相互作用指数分析,讨论了构象异构体稳定性的计算细节。
更新日期:2020-05-04
中文翻译:
N-(噻唑-2-基)苯甲酰胺构象偏好的计算研究
在某些生物活性化合物中发现了N-(噻唑-2-基)苯甲酰胺1亚结构。在某些蛋白质/配体共晶体中,1部分采用的构象异构体中酰胺O和噻唑S原子紧密相连。实际上,在结晶结构1中,O-S距离比范德华半径更短。尽管自然键轨道分析发现O和S原子之间的稳定相互作用较弱,但酰胺N–H和噻唑N原子之间有吸引力的偶极-偶极相互作用似乎起着更重要的作用。此外,发现二聚体形式的分子内OH氢键对1的构象偏好具有重要作用。使用分子中原子的量子理论,自然键轨道(NBO)和非共价相互作用指数分析,讨论了构象异构体稳定性的计算细节。








































 京公网安备 11010802027423号
京公网安备 11010802027423号