当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electronic Structure and Optical Properties of Vacancy-Ordered Double Perovskites Cs2Pd BrxCl6–x by First-Principles Calculation
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-04-30 , DOI: 10.1021/acs.jpcc.0c00137 Zi-Yan Wang 1 , Yi Chen 1 , Chongyang Zhang 2 , Dan Wang 1 , Pei Liang 1 , Hong Zhang 1 , Rong-Jun Xie 2 , Le Wang 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-04-30 , DOI: 10.1021/acs.jpcc.0c00137 Zi-Yan Wang 1 , Yi Chen 1 , Chongyang Zhang 2 , Dan Wang 1 , Pei Liang 1 , Hong Zhang 1 , Rong-Jun Xie 2 , Le Wang 1
Affiliation
|
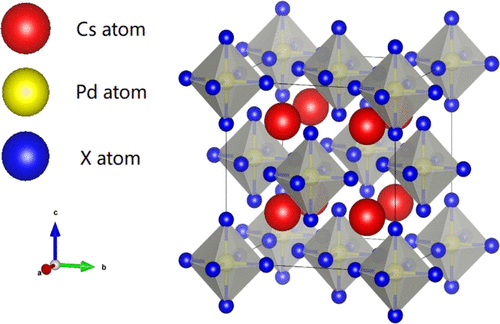
Vacancy-ordered double perovskites, having a great application in optoelectronic and thermoelectric devices, are variants of metal-halide perovskites. The PBE and HSE06 functionals for the first-principles calculation were used to investigate the electronic structures, optical properties, possible intrinsic defects, and formation energies of the vacancy-ordered double perovskites Cs2PdBrxCl6–x (x = 1, 2, 3, ..., 6). All the compounds had a direct band gap, and the band gap increased from 1.566 eV for Cs2PdBr6 to 2.675 eV for Cs2PdCl6 when Br atoms are replaced with Cl atoms. The absorption of these compounds blue-shifted with increasing Cl:Br ratio, and higher absorption coefficients can thus be acquired in the ultraviolet region. The defect calculations showed that the formation energies of VCs, VPd, and CsPd were low, and the Fermi level was pinned near the valence band maximum. The results indicate that vacancy-ordered double perovskites are ideal candidates for lead-free solar cells with high conversion efficiencies.
中文翻译:

空位有序双钙钛矿Cs 2 Pd Br x Cl 6– x的电子结构和光学性质的第一性原理计算
空缺有序的钙钛矿是金属卤化物钙钛矿的变体,在光电和热电设备中有很大的应用。用于第一性原理计算的PBE和HSE06功能用于研究空位有序双钙钛矿Cs 2 PdBr x Cl 6– x(x = 1,2)的电子结构,光学性质,可能的固有缺陷和形成能,3,...,6)。所有化合物均具有直接带隙,并且带隙从Cs 2 PdBr 6的1.566 eV增加到Cs 2 PdCl 6的2.675 eV当Br原子被Cl原子取代时。这些化合物的吸收随着Cl:Br比的增加而蓝移,因此可以在紫外线区域获得更高的吸收系数。缺陷计算表明,V Cs,V Pd和Cs Pd的形成能较低,费米能级固定在价带最大值附近。结果表明,空位排序的双重钙钛矿是具有高转换效率的无铅太阳能电池的理想候选者。
更新日期:2020-06-18
中文翻译:
空位有序双钙钛矿Cs 2 Pd Br x Cl 6– x的电子结构和光学性质的第一性原理计算
空缺有序的钙钛矿是金属卤化物钙钛矿的变体,在光电和热电设备中有很大的应用。用于第一性原理计算的PBE和HSE06功能用于研究空位有序双钙钛矿Cs 2 PdBr x Cl 6– x(x = 1,2)的电子结构,光学性质,可能的固有缺陷和形成能,3,...,6)。所有化合物均具有直接带隙,并且带隙从Cs 2 PdBr 6的1.566 eV增加到Cs 2 PdCl 6的2.675 eV当Br原子被Cl原子取代时。这些化合物的吸收随着Cl:Br比的增加而蓝移,因此可以在紫外线区域获得更高的吸收系数。缺陷计算表明,V Cs,V Pd和Cs Pd的形成能较低,费米能级固定在价带最大值附近。结果表明,空位排序的双重钙钛矿是具有高转换效率的无铅太阳能电池的理想候选者。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号