当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
块状ZnX(X = O,S,Se,Te),ZnF2和ZnO / ZnF2的结构和电子性质:PBE,PBE + U和混合HSE功能内的DFT研究。
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-05-04 , DOI: 10.1021/acs.jpca.9b11415 Efracio Mamani Flores 1, 2 , Mário L Moreira 1 , Maurício Jeomar Piotrowski 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-05-04 , DOI: 10.1021/acs.jpca.9b11415 Efracio Mamani Flores 1, 2 , Mário L Moreira 1 , Maurício Jeomar Piotrowski 1
Affiliation
|
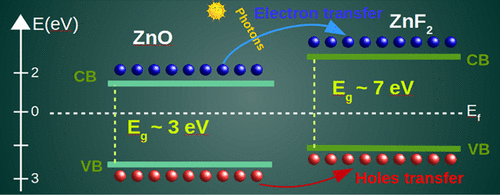
在这里,我们研究了本体ZnX(X = O,S,Se,Te)和ZnF2系统的晶体结构,这是了解ZnX和ZnO / ZnF2界面等基于Zn的系统的结构的第一步。对于可能的技术应用的重要性。此外,基于密度泛函理论(DFT)计算的充分方法学描述是必要的。众所周知,基于局部或半局部交换相关函数的普通DFT计算无法为这些系统描述正确的带隙能量,而诸如基于混合函数的非局部方法可以补偿带隙的低估。为了促进评估,在半本地Perdew-Burke-Ernzerhof(PBE)和两个非本地功能中进行了DFT研究,Heyd-Scuseria-Ernzerhof(HSE)和PBE + U功能的混合体。我们的结果证实,与实验值相比,PBE低估了ZnX化合物的能带隙值,从33.0到42.8%。应用混合HSE泛函,我们获得了与非局部精确交换的分离范围有关的带隙依赖性,通常可以减小带隙误差并改善晶格常数描述。另外,使用PBE + U方法,我们研究了Zn d态的定位及其对ZnX和ZnF2中带隙的影响。我们发现带隙随着Hubbard参数的增加而增加,这引入了针对Zn 3d态的现场库仑校正。在相同的上下文中,突出显示了包括O 2p状态(和X p状态)的Hubbard校正的相关性。从而,考虑到PBE + U,相对于实验值,例如ZnO带隙的误差降低到5.1%。最后,发现ZnO-12L / ZnF2-4L超晶格表现出常规的电子特性,例如低的基带隙,小于任何一种母体材料。我们的第一性原理计算表明,带电层的意外减小是由导电层引起的,这些导电层倾向于穿透界面并减小带隙,从而导致载流子通过界面传输到ZnF2,即使带隙较大对于电荷转移,对于光伏应用可能是有趣的。例如低的基带隙,小于任何一种母体材料。我们的第一性原理计算表明,带隙的意外减少是由倾向于穿透界面并减小带隙的导电层引起的,从而导致载流子通过界面传输到ZnF2,即使带隙较大对于电荷转移,对于光伏应用可能是有趣的。例如低的基带隙,小于任何一种母体材料。我们的第一性原理计算表明,带隙的意外减少是由倾向于穿透界面并减小带隙的导电层引起的,从而导致载流子通过界面传输到ZnF2,即使带隙较大对于电荷转移,对于光伏应用可能是有趣的。

"点击查看英文标题和摘要"
更新日期:2020-04-24
"点击查看英文标题和摘要"





























 京公网安备 11010802027423号
京公网安备 11010802027423号