当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Neural Network Interatomic Potential for Predicting the Formation of Planar Defect in Nanocrystal
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-04-21 , DOI: 10.1021/acs.jpcc.9b11698 Kyoungmin Min 1 , Eunseog Cho 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2020-04-21 , DOI: 10.1021/acs.jpcc.9b11698 Kyoungmin Min 1 , Eunseog Cho 1
Affiliation
|
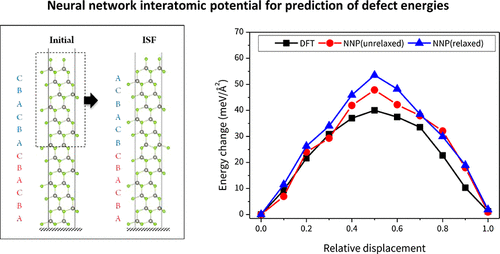
Recent advances in the development of interatomic potential using neural networks have proven that its accuracy reaches that of first-principles calculations but with considerably reduced computational cost. In this study, we successfully implement a neural network to construct the interatomic potential of the ZnSe structure by training its potential energy surface results obtained from density functional theory (DFT) calculations. The developed potential is used for molecular dynamics simulations and its accuracy lies within an error of 6% on average from the DFT results for predicting the total energy on pristine and defective bulk structures, slab, and cluster structures of ZnSe. The prediction accuracy is also demonstrated considering the lattice constant and mechanical properties of the pristine bulk structure. To demonstrate its transferability further, a neural network potential is constructed to predict the formation energy of planar defects (stacking fault and twin boundary) in the slab and the nanocrystal structures, and it precisely reproduces the order of stability for each defect type. These results reveal that the neural-network-based interatomic potential can be used to revolutionize atomistic simulations by significantly saving the computation time while maintaining accuracy comparable to that of the DFT.
中文翻译:

神经网络原子间势用于预测纳米晶体中平面缺陷的形成
使用神经网络开发原子间势的最新进展已证明其精度可以达到第一性原理的计算,但计算成本却大大降低。在这项研究中,我们通过训练从密度泛函理论(DFT)计算获得的ZnSe结构的势能面结果,成功地实现了神经网络来构造ZnSe结构的原子间势。所开发的电势用于分子动力学模拟,其准确性与DFT结果的平均误差在6%以内,用于预测ZnSe的原始和有缺陷的块状结构,平板和簇结构上的总能量。还考虑了原始块状结构的晶格常数和机械性能,证明了预测精度。为了进一步证明其可传递性,构建了一个神经网络电势来预测平板和纳米晶体结构中平面缺陷(堆叠缺陷和孪晶边界)的形成能,并精确地再现每种缺陷类型的稳定性顺序。这些结果表明,基于神经网络的原子间电势可以显着节省计算时间,同时保持与DFT相当的精度,从而可以使原子模拟发生革命性变化。
更新日期:2020-04-24
中文翻译:
神经网络原子间势用于预测纳米晶体中平面缺陷的形成
使用神经网络开发原子间势的最新进展已证明其精度可以达到第一性原理的计算,但计算成本却大大降低。在这项研究中,我们通过训练从密度泛函理论(DFT)计算获得的ZnSe结构的势能面结果,成功地实现了神经网络来构造ZnSe结构的原子间势。所开发的电势用于分子动力学模拟,其准确性与DFT结果的平均误差在6%以内,用于预测ZnSe的原始和有缺陷的块状结构,平板和簇结构上的总能量。还考虑了原始块状结构的晶格常数和机械性能,证明了预测精度。为了进一步证明其可传递性,构建了一个神经网络电势来预测平板和纳米晶体结构中平面缺陷(堆叠缺陷和孪晶边界)的形成能,并精确地再现每种缺陷类型的稳定性顺序。这些结果表明,基于神经网络的原子间电势可以显着节省计算时间,同时保持与DFT相当的精度,从而可以使原子模拟发生革命性变化。






























 京公网安备 11010802027423号
京公网安备 11010802027423号