Nature Chemistry ( IF 19.2 ) Pub Date : 2020-03-30 , DOI: 10.1038/s41557-020-0438-z
Weijie Chen 1 , Anirudra Paul 1 , Khalil A Abboud 2 , Daniel Seidel 1
|
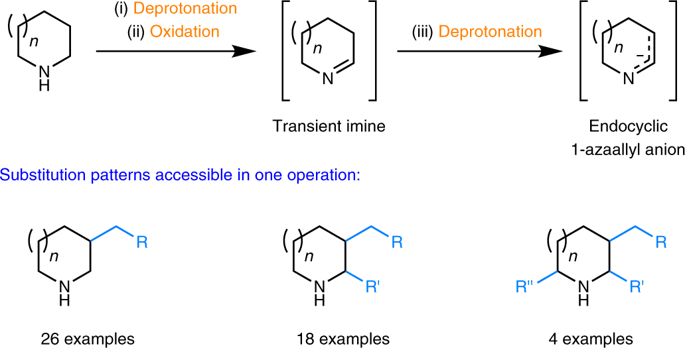
The synthesis of valuable bioactive alicyclic amines containing variable substituents in multiple ring positions typically relies on multistep synthetic sequences that frequently require the introduction and subsequent removal of undesirable protecting groups. Although a vast number of studies have aimed to simplify access to such materials through the C–H bond functionalization of feedstock alicyclic amines, the simultaneous introduction of more than one substituent to unprotected amines has never been accomplished. Here we report an advance in C–H bond functionalization methodology that enables the introduction of up to three substituents in a single operation. Lithiated amines are first exposed to a ketone oxidant, generating transient imines that are subsequently converted to endocyclic 1-azaallyl anions, which can be processed further to furnish β-substituted, α,β-disubstituted, or α,β,α′-trisubstituted amines. This study highlights the unique utility of in situ-generated endocyclic 1-azaallyl anions, elusive intermediates in synthetic chemistry.

中文翻译:
在未保护的脂环族胺中多个CH键的快速官能化。
在多个环位置含有可变取代基的有价值的生物活性脂环族胺的合成通常依赖于多步合成序列,该序列通常需要引入并随后除去不希望的保护基团。尽管大量研究旨在通过原料脂环族胺的C–H键官能化来简化此类材料的获得,但是从未将同时取代多个取代基的胺引入未保护的胺中从未实现。在这里,我们报告了CH键官能化方法的一项进展,该方法可在一次操作中引入多达三个取代基。首先将锂化的胺暴露于酮氧化剂,生成瞬态亚胺,随后将其转化为环内1-氮杂烯丙基阴离子,可以进一步加工以提供β-取代的,α,β-二取代的或α,β,α'-三取代的胺。这项研究突出了原位生成的环内1-氮杂烯丙基阴离子(合成化学中难以捉摸的中间体)的独特用途。






































 京公网安备 11010802027423号
京公网安备 11010802027423号