Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy ( IF 4.3 ) Pub Date : 2020-03-05 , DOI: 10.1016/j.saa.2020.118216 Francisco Colmenero , Jakub Plášil , Pavel Škácha
|
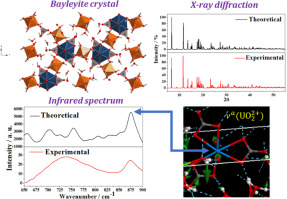
Bayleyite is a highly hydrated uranyl tricarbonate mineral containing eighteen water molecules per formula unit. Due to this large water content, the correct description of its crystal structure is a great challenge for the first principles solid state methodology. In this work, the crystal structure, hydrogen bonding, mechanical properties and infrared spectrum of bayleyite, Mg2[UO2(CO3)3] · 18 H2O, have been investigated by means of Periodic Density Functional Theory methods using plane wave basis sets and pseudopotentials. The computed unit-cell parameters, interatomic distances, hydrogen bonding network geometry and the X-ray powder diffraction pattern of bayleyite reproduce successfully the experimental data, thus confirming the crystal structure determined from X-ray diffraction data. From the energy-optimized structure, the elastic properties and infrared spectrum have been determined using theoretical methods. The calculated elastic properties include the bulk modulus and its pressure derivatives, the Young and shear moduli, the Poisson ratio and the ductility, hardness and anisotropy indices. Bayleyite is shown to be a very isotropic ductile mineral possessing a bulk modulus of B ~28 GPa. The infrared spectrum of bayleyite is obtained experimentally from a natural mineral sample from the Jáchymov ore district, Czech Republic, and determined employing density functional perturbation theory. Since both spectra show a high degree of consistence, the bands in the observed spectrum are assigned using the theoretical methodology. The atomic vibrational motions localized in the uranyl tricarbonate units are described in detail, using appropriate normal coordinate analyses based on accurate vibrational computations, since the vibrational normal modes have not been hitherto studied for any uranyl tricarbonate mineral.
中文翻译:
铀酰三碳酸铀酰八水合物矿物,贝利石:周期性DFT研究其晶体结构,氢键,机械性能和红外光谱
贝利石是高度水合的三碳酸铀酰矿物质,每个配方单位包含18个水分子。由于水含量高,因此对于其晶体结构的正确描述对于第一原理固态方法学是一个巨大的挑战。在这项工作中,贝利石Mg 2 [UO 2(CO 3)3 ]·18 H 2的晶体结构,氢键,力学性能和红外光谱O,已经通过使用平面波基集和伪势的周期性密度泛函理论方法进行了研究。计算出的晶胞参数,原子间距离,氢键网络的几何形状以及贝叶石的X射线粉末衍射图成功地再现了实验数据,从而确认了由X射线衍射数据确定的晶体结构。从能量优化结构,已经使用理论方法确定了弹性和红外光谱。计算的弹性特性包括体积模量及其压力导数,杨氏模量和剪切模量,泊松比以及延展性,硬度和各向异性指数。贝利石被证明是一种非常各向同性的延性矿物,具有的体积模量为B〜28 GPa。贝利石的红外光谱是通过实验从捷克共和国贾奇莫夫矿区的天然矿物样品中获得的,并采用密度泛函微扰理论进行了测定。由于两个光谱均显示高度一致性,因此使用理论方法对观察光谱中的谱带进行分配。由于迄今尚未对任何三碳酸铀基酯的振动标准振型进行研究,因此基于精确的振动计算,使用适当的法向坐标分析,详细描述了三碳酸铀基酯单元中的原子振动运动。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号