当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unveiling the Active Structure of Single Nickel Atom Catalysis: Critical Roles of Charge Capacity and Hydrogen Bonding
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-03-03 , DOI: 10.1021/jacs.9b13872 Xunhua Zhao 1 , Yuanyue Liu 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-03-03 , DOI: 10.1021/jacs.9b13872 Xunhua Zhao 1 , Yuanyue Liu 1
Affiliation
|
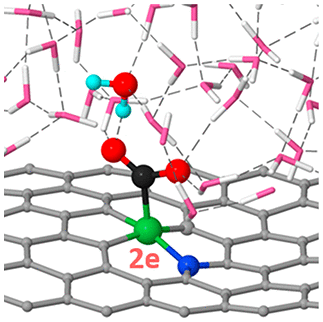
Single nickel atom embedded in graphene is one of the most representative single atom catalysts, and has a high activity and selectivity for electrochemical CO2 reduction (CO2R) to CO. However, the catalytic origin, especially the coordination structure of Ni, remains highly puzzling, as previous density-functional-theory (DFT) calculations show all the possible structures should be inactive and/or non-selective. Here using ab-initio molecular dynamics (AIMD) and "slow-growth" sampling approach to evaluate the reaction kinetic barriers, we show that the charge capacity (of the site) and hydrogen bonding (with the intermediates), which were neglected/oversimplified in previous DFT calculations, play crucial roles, and including their effects can resolve the catalytic origin. Particularly, a high charge capacity allows the catalytic site to carry more charges than required for electrochemical step, lowering the electrochemical barrier; the hydrogen bonding promotes the reaction that produces polar intermediates, by stabilizing the intermediates and facilitating the H transfer from water, explaining the high selectivity for CO2R over hydrogen evolution reaction. Consequently, we find a hybrid coordination environment (with one nitrogen and three carbon atoms) for Ni atom is most active and selective for CO2R. Our work not only explains a long-standing puzzle for an important catalyst, but also highlights the crucial roles of charge capacity and hydrogen bonding, which can help elucidate the mechanisms of other heterogeneous electrocatalysts in aqueous solution and enable more effective catalyst design.
中文翻译:

揭示单镍原子催化的活性结构:电荷容量和氢键的关键作用
嵌入石墨烯中的单个镍原子是最具代表性的单原子催化剂之一,对电化学 CO2 还原 (CO2R) 至 CO 具有很高的活性和选择性。 然而,催化起源,尤其是 Ni 的配位结构,仍然非常令人费解,因为之前的密度泛函理论 (DFT) 计算表明所有可能的结构都应该是非活性和/或非选择性的。在这里使用 ab-initio 分子动力学 (AIMD) 和“缓慢增长”采样方法来评估反应动力学障碍,我们表明(位点的)和氢键(与中间体)的电荷容量被忽略/过度简化在之前的 DFT 计算中,起着至关重要的作用,包括它们的影响可以解决催化起源。特别,高充电容量允许催化位点携带比电化学步骤所需的更多的电荷,从而降低电化学势垒;氢键通过稳定中间体和促进水中的 H 转移来促进产生极性中间体的反应,解释了 CO2R 对析氢反应的高选择性。因此,我们发现 Ni 原子的混合配位环境(具有一个氮和三个碳原子)对 CO2R 最具活性和选择性。我们的工作不仅解释了一个重要催化剂的长期难题,而且突出了电荷容量和氢键的关键作用,这有助于阐明水溶液中其他非均相电催化剂的机制,并实现更有效的催化剂设计。
更新日期:2020-03-03
中文翻译:
揭示单镍原子催化的活性结构:电荷容量和氢键的关键作用
嵌入石墨烯中的单个镍原子是最具代表性的单原子催化剂之一,对电化学 CO2 还原 (CO2R) 至 CO 具有很高的活性和选择性。 然而,催化起源,尤其是 Ni 的配位结构,仍然非常令人费解,因为之前的密度泛函理论 (DFT) 计算表明所有可能的结构都应该是非活性和/或非选择性的。在这里使用 ab-initio 分子动力学 (AIMD) 和“缓慢增长”采样方法来评估反应动力学障碍,我们表明(位点的)和氢键(与中间体)的电荷容量被忽略/过度简化在之前的 DFT 计算中,起着至关重要的作用,包括它们的影响可以解决催化起源。特别,高充电容量允许催化位点携带比电化学步骤所需的更多的电荷,从而降低电化学势垒;氢键通过稳定中间体和促进水中的 H 转移来促进产生极性中间体的反应,解释了 CO2R 对析氢反应的高选择性。因此,我们发现 Ni 原子的混合配位环境(具有一个氮和三个碳原子)对 CO2R 最具活性和选择性。我们的工作不仅解释了一个重要催化剂的长期难题,而且突出了电荷容量和氢键的关键作用,这有助于阐明水溶液中其他非均相电催化剂的机制,并实现更有效的催化剂设计。




























 京公网安备 11010802027423号
京公网安备 11010802027423号