Nature Medicine ( IF 58.7 ) Pub Date : 2020-02-10 , DOI: 10.1038/s41591-019-0729-3 Pedro Milanez-Almeida 1, 2 , Andrew J Martins 3 , Ronald N Germain 1 , John S Tsang 2, 3
|
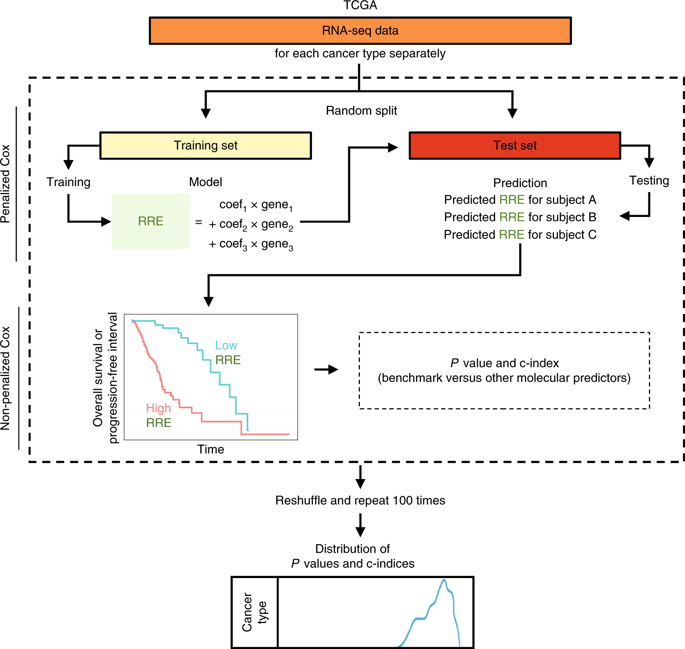
Disrupted molecular pathways are often robustly associated with disease outcome in cancer1,2,3. Although biologically informative transcriptional pathways can be revealed by RNA sequencing (RNA-seq) at up to hundreds of folds reduction in conventionally used coverage4,5,6, it remains unknown how low-depth sequencing datasets perform in the challenging context of developing transcriptional signatures to predict clinical outcomes. Here we assessed the possibility of cancer prognosis with shallow tumor RNA-seq, which would potentially enable cost-effective assessment of much larger numbers of samples for deeper biological and predictive insights. By statistically modeling the relative risk of an adverse outcome for thousands of subjects in The Cancer Genome Atlas7,8,9,10,11,12,13, we present evidence that subsampled tumor RNA-seq data with a few hundred thousand reads per sample provide sufficient information for outcome prediction in several types of cancer. Analysis of predictive models revealed robust contributions from pathways known to be associated with outcomes. Our findings indicate that predictive models of outcomes in cancer may be developed with dramatically increases in sample numbers at low cost, thus potentially enabling the development of more realistic predictive models that incorporate diverse variables and their interactions. This strategy could also be used, for example, in longitudinal analysis of multiple regions of a tumor alongside treatment for quantitative modeling and prediction of outcome in personalized oncology.
中文翻译:
浅层肿瘤 RNA 测序的癌症预后
被破坏的分子途径通常与癌症1,2,3的疾病结果密切相关。尽管通过 RNA 测序 (RNA-seq) 可以揭示具有生物学信息的转录途径,在常规使用的覆盖范围内减少了数百倍4,5,6,但在开发转录的具有挑战性的背景下,低深度测序数据集的表现如何仍然未知预测临床结果的特征。在这里,我们使用浅层肿瘤 RNA-seq 评估了癌症预后的可能性,这将有可能对大量样本进行具有成本效益的评估,以获得更深入的生物学和预测见解。通过对癌症基因组图谱中数千名受试者的不良结果的相对风险进行统计建模7,8,9,10,11,12,13,我们提供的证据表明,每个样本有几十万个读数的二次采样肿瘤 RNA-seq 数据为几种癌症的结果预测提供了足够的信息。预测模型的分析揭示了已知与结果相关的途径的强大贡献。我们的研究结果表明,癌症结果的预测模型可以通过以低成本显着增加样本数量来开发,从而有可能开发更现实的预测模型,其中包含不同的变量及其相互作用。例如,该策略还可用于肿瘤多个区域的纵向分析以及个性化肿瘤学结果的定量建模和预测治疗。










































 京公网安备 11010802027423号
京公网安备 11010802027423号