当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of chemical nature of atoms on the electronic, dielectric, and dynamical properties of ABX3 halide perovskite
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-02-03 , DOI: 10.1002/qua.26172 Raouia Ben Sadok 1 , Neculai Plugaru 2 , Anca Birsan 2 , Victor Kuncser 2 , Dalila Hammoutène 1
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2020-02-03 , DOI: 10.1002/qua.26172 Raouia Ben Sadok 1 , Neculai Plugaru 2 , Anca Birsan 2 , Victor Kuncser 2 , Dalila Hammoutène 1
Affiliation
|
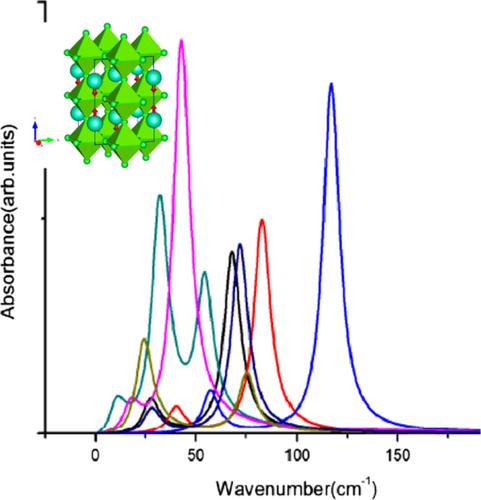
First‐principles calculations within density functional theory were performed on a series of halide perovskite compounds ABX3(A: Cs or Rb; B:Pb or Sn). Their electronic structure, lattice dynamics, and dielectric properties were studied in relationship with the change in atom species at each one of the three inequivalent crystallographic sites, to explain the origin of these properties. Thus, the variation of the bandgap with the overlap between the B cation lone pair and the electronic states of halide atoms, as well as with the distortion of the BX6 octahedra network is discussed. It is shown that the vibrational modes, phonon frequencies, atomic displacements, and the possible ferroelectric instability in these compounds are dependent on masses of atoms, volume of AX12 polyhedron, as well as on streoactivity of Pb lone pair. Also, the Born effective charges, dielectric constant, spontaneous polarization, and infrared spectra are calculated. The relation between these dielectric properties and the ions dynamics is discussed.
中文翻译:

原子的化学性质对ABX3卤化物钙钛矿的电子,介电和动力学性质的影响
密度泛函理论中的第一性原理计算是对一系列卤化物钙钛矿化合物ABX 3(A:Cs或Rb;B:Pb或Sn)进行的。研究了它们的电子结构,晶格动力学和介电特性,并与三个不等价晶体学位置中每一个的原子种类的变化相关,以解释这些特性的起源。因此,带隙的变化与B阳离子孤对和卤原子原子的电子态之间的重叠以及BX 6的畸变有关讨论了八面体网络。结果表明,这些化合物的振动模式,声子频率,原子位移和可能的铁电不稳定性取决于原子质量,AX 12多面体的体积以及Pb孤对的结构活性。而且,计算出了玻恩有效电荷,介电常数,自发极化和红外光谱。讨论了这些介电特性和离子动力学之间的关系。
更新日期:2020-04-06
中文翻译:
原子的化学性质对ABX3卤化物钙钛矿的电子,介电和动力学性质的影响
密度泛函理论中的第一性原理计算是对一系列卤化物钙钛矿化合物ABX 3(A:Cs或Rb;B:Pb或Sn)进行的。研究了它们的电子结构,晶格动力学和介电特性,并与三个不等价晶体学位置中每一个的原子种类的变化相关,以解释这些特性的起源。因此,带隙的变化与B阳离子孤对和卤原子原子的电子态之间的重叠以及BX 6的畸变有关讨论了八面体网络。结果表明,这些化合物的振动模式,声子频率,原子位移和可能的铁电不稳定性取决于原子质量,AX 12多面体的体积以及Pb孤对的结构活性。而且,计算出了玻恩有效电荷,介电常数,自发极化和红外光谱。讨论了这些介电特性和离子动力学之间的关系。





























 京公网安备 11010802027423号
京公网安备 11010802027423号