当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Halogen Bonds in Ligand-Protein Systems: Molecular Orbital Theory for Drug Design.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-31 , DOI: 10.1021/acs.jcim.9b00946 Enrico Margiotta 1, 2 , Stephanie C C van der Lubbe 1 , Lucas de Azevedo Santos 1, 3 , Gabor Paragi 1, 4, 5 , Stefano Moro 2 , F Matthias Bickelhaupt 1, 6 , Célia Fonseca Guerra 1, 7
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-31 , DOI: 10.1021/acs.jcim.9b00946 Enrico Margiotta 1, 2 , Stephanie C C van der Lubbe 1 , Lucas de Azevedo Santos 1, 3 , Gabor Paragi 1, 4, 5 , Stefano Moro 2 , F Matthias Bickelhaupt 1, 6 , Célia Fonseca Guerra 1, 7
Affiliation
|
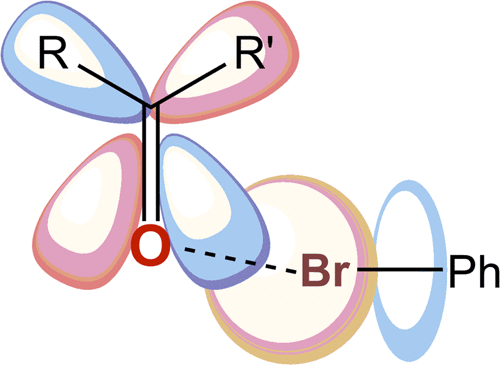
Halogen bonds are highly important in medicinal chemistry as halogenation of drugs, generally, improves both selectivity and efficacy toward protein active sites. However, accurate modeling of halogen bond interactions remains a challenge, since a thorough theoretical investigation of the bonding mechanism, focusing on the realistic complexity of drug-receptor systems, is lacking. Our systematic quantum-chemical study on ligand/peptide-like systems reveals that halogen bonding is driven by the same bonding interactions as hydrogen bonding. Besides the electrostatic and the dispersion interactions, our bonding analyses, based on quantitative Kohn-Sham molecular orbital theory together with energy decomposition analysis, reveal that donor-acceptor interactions and steric repulsion between the occupied orbitals of the halogenated ligand and the protein need to be considered more carefully within the drug design process.
中文翻译:

配体-蛋白质系统中的卤素键:药物设计的分子轨道理论。
卤素键在药物化学中非常重要,因为药物的卤化通常会提高对蛋白质活性位点的选择性和功效。然而,由于缺乏对键合机理进行透彻的理论研究,而侧重于药物受体系统的实际复杂性,因此,对卤素键相互作用的准确建模仍然是一个挑战。我们对配体/肽样系统的系统量子化学研究表明,卤素键是由与氢键相同的键相互作用驱动的。除了静电和色散相互作用外,我们的键合分析基于定量的Kohn-Sham分子轨道理论以及能量分解分析,
更新日期:2020-01-31
中文翻译:
配体-蛋白质系统中的卤素键:药物设计的分子轨道理论。
卤素键在药物化学中非常重要,因为药物的卤化通常会提高对蛋白质活性位点的选择性和功效。然而,由于缺乏对键合机理进行透彻的理论研究,而侧重于药物受体系统的实际复杂性,因此,对卤素键相互作用的准确建模仍然是一个挑战。我们对配体/肽样系统的系统量子化学研究表明,卤素键是由与氢键相同的键相互作用驱动的。除了静电和色散相互作用外,我们的键合分析基于定量的Kohn-Sham分子轨道理论以及能量分解分析,
































 京公网安备 11010802027423号
京公网安备 11010802027423号