Journal of Catalysis ( IF 6.5 ) Pub Date : 2020-01-17 , DOI: 10.1016/j.jcat.2019.12.034
Nirala Singh , Udishnu Sanyal , Griffin Ruehl , Kelsey A. Stoerzinger , Oliver Y. Gutiérrez , Donald M. Camaioni , John L. Fulton , Johannes A. Lercher , Charles T. Campbell
|
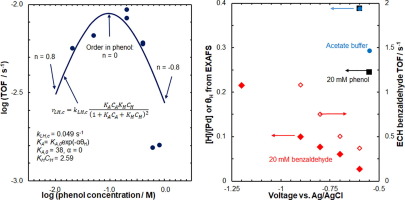
The mechanisms of aqueous-phase thermal catalytic hydrogenation (TCH) and electrocatalytic hydrogenation (ECH) of organic molecules over Pt group metals are not as well-understood as gas-phase thermal catalytic hydrogenation. In gas-phase, the reactions generally occur via a Langmuir-Hinshelwood mechanism with adsorbed hydrogen adding to adsorbed organics. Here, we show that the rates, reaction orders and activation energies for TCH and ECH of phenol and benzaldehyde on Pd, Pt, and Rh can be explained with a simple kinetic model based on similar Langmuir-Hinshelwood mechanisms. For Pt/C, the adsorption equilibrium constants for the organics needed to fit the rate data are consistent with independently-measured values, provided we assume that the rates are dominated by (1 1 1)-like sites, in agreement with reported particle size effects. The reaction rate of Pd in the ECH of benzaldehyde increases with the surface hydrogen coverage. The state of Pd during ECH of phenol and benzaldehyde are very different, with a high concentration of adsorbed H in the presence of phenol, but not in the presence of benzaldehyde, consistent with benzaldehyde’s stronger binding to the surface. In consequence, Pd is converted to β-PdHx during the hydrogenation of phenol but not benzaldehyde. This is proposed to explain the much lower activity of Pd for hydrogenation of phenol compared to benzaldehyde. The measured low coverage of H on Pd in the presence of benzaldehyde is in agreement with the high selectivity/Faradaic efficiency of protons to benzaldehyde hydrogenation to benzyl alcohol. The decrease in apparent activation energy for ECH versus TCH can also be understood within this same kinetic model. The combination of ECH and TCH kinetics and spectroscopy has, thus, allowed to deduce a microkinetic model for these hydrogenation reactions.
中文翻译:
铂族金属上苯酚和苯甲醛的水相催化和电催化加氢
铂族金属上有机分子的水相热催化加氢(TCH)和电催化加氢(ECH)的机理不像气相热催化加氢那样容易理解。在气相中,反应通常通过Langmuir-Hinshelwood机理发生,将吸附的氢添加到吸附的有机物中。在这里,我们表明,酚和苯甲醛在Pd,Pt和Rh上的TCH和ECH的速率,反应顺序和活化能可以通过基于相似Langmuir-Hinshelwood机理的简单动力学模型来解释。对于Pt / C,拟合速率数据所需的有机物的吸附平衡常数与独立测量的值一致,前提是我们假设速率受(1 1 1)样位点支配,并与所报告的粒径一致效果。苯甲醛ECH中Pd的反应速率随表面氢的覆盖率而增加。苯酚和苯甲醛ECH期间Pd的状态有很大不同,在苯酚存在下高浓度的H吸附,但在苯甲醛不存在时吸附P的状态高,这与苯甲醛与表面的牢固结合相一致。结果,Pd转化为β-PdHx在苯酚而不是苯甲醛的氢化过程中。提出这是为了解释与苯甲醛相比,钯对苯酚氢化的活性要低得多。在苯甲醛存在下测得的H在Pd上的低覆盖率与质子对苯甲醛加氢制苯甲醇的高选择性/法拉第效率一致。在相同的动力学模型中,也可以理解ECH与TCH的表观活化能的降低。因此,ECH和TCH动力学和光谱学的结合可以推导这些氢化反应的微动力学模型。
































 京公网安备 11010802027423号
京公网安备 11010802027423号