当前位置:
X-MOL 学术
›
J. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Anharmonic modeling of the conformation-specific IR spectra of ethyl, n-propyl, and n-butylbenzene
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2016-06-14 12:25:21 , DOI: 10.1063/1.4953181 Daniel P. Tabor 1 , Daniel M. Hewett 2 , Sebastian Bocklitz 2 , Joseph A. Korn 2 , Anthony J. Tomaine 2 , Arun K. Ghosh 2 , Timothy S. Zwier 2 , Edwin L. Sibert 1
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2016-06-14 12:25:21 , DOI: 10.1063/1.4953181 Daniel P. Tabor 1 , Daniel M. Hewett 2 , Sebastian Bocklitz 2 , Joseph A. Korn 2 , Anthony J. Tomaine 2 , Arun K. Ghosh 2 , Timothy S. Zwier 2 , Edwin L. Sibert 1
Affiliation
|
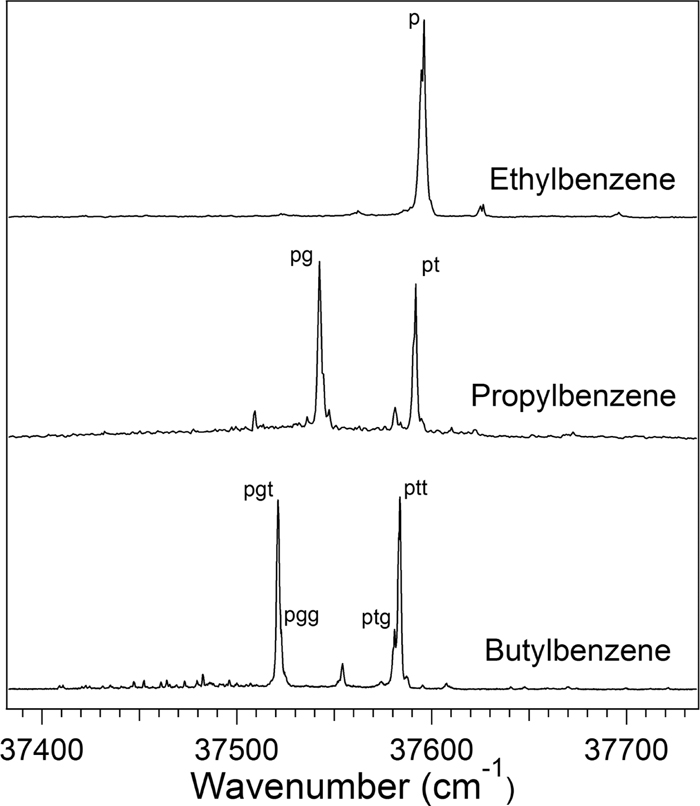
Conformation-specific UV-IR double resonance spectra are presented for ethyl, n-propyl, and n-butylbenzene. With the aid of a local mode Hamiltonian that includes the effects of stretch-scissor Fermi resonance, the spectra can be accurately modeled for specific conformers. These molecules allow for further development of a first principles method for calculating alkyl stretch spectra. Across all chain lengths, certain dihedral patterns impart particular spectral motifs at the quadratic level. However, the anharmonic contributions are consistent from molecule to molecule and conformer to conformer. This transferability of anharmonicities allows for the Hamiltonian to be constructed from only a harmonic frequency calculation, reducing the cost of the model. The phenyl ring alters the frequencies of the CH2 stretches by about 15 cm−1 compared to their n-alkane counterparts in trans configurations. Conformational changes in the chain can lead to shifts in frequency of up to 30 cm−1.
中文翻译:

乙基,正丙基和正丁基苯的构象特定红外光谱的非谐建模
给出了乙基,正丙基和正丁基的构象特异性UV-IR双共振光谱丁基苯。借助包含拉伸剪刀费米共振效应的局部模式哈密顿量,可以针对特定的构象异构体准确建模光谱。这些分子允许进一步发展用于计算烷基拉伸光谱的第一原理方法。在所有链长上,某些二面体图案在二次水平上赋予特定的光谱图案。但是,非谐贡献在分子之间和构象异构体之间是一致的。非谐性的这种可传递性允许仅通过谐波频率计算来构造哈密顿量,从而降低了模型的成本。与n相比,苯环将CH 2拉伸的频率改变了约15 cm -1反式构型中的-烷烃对应物。链中的构象变化可能导致频率变化最多30 cm -1。
更新日期:2016-06-15
中文翻译:
乙基,正丙基和正丁基苯的构象特定红外光谱的非谐建模
给出了乙基,正丙基和正丁基的构象特异性UV-IR双共振光谱丁基苯。借助包含拉伸剪刀费米共振效应的局部模式哈密顿量,可以针对特定的构象异构体准确建模光谱。这些分子允许进一步发展用于计算烷基拉伸光谱的第一原理方法。在所有链长上,某些二面体图案在二次水平上赋予特定的光谱图案。但是,非谐贡献在分子之间和构象异构体之间是一致的。非谐性的这种可传递性允许仅通过谐波频率计算来构造哈密顿量,从而降低了模型的成本。与n相比,苯环将CH 2拉伸的频率改变了约15 cm -1反式构型中的-烷烃对应物。链中的构象变化可能导致频率变化最多30 cm -1。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号