当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Soft-Sphere Continuum Solvation in Electronic-Structure Calculations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-07-03 00:00:00 , DOI: 10.1021/acs.jctc.7b00375
Giuseppe Fisicaro 1 , Luigi Genovese 2 , Oliviero Andreussi 3, 4 , Sagarmoy Mandal 5 , Nisanth N. Nair 5 , Nicola Marzari 4 , Stefan Goedecker 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-07-03 00:00:00 , DOI: 10.1021/acs.jctc.7b00375
Giuseppe Fisicaro 1 , Luigi Genovese 2 , Oliviero Andreussi 3, 4 , Sagarmoy Mandal 5 , Nisanth N. Nair 5 , Nicola Marzari 4 , Stefan Goedecker 1
Affiliation
|
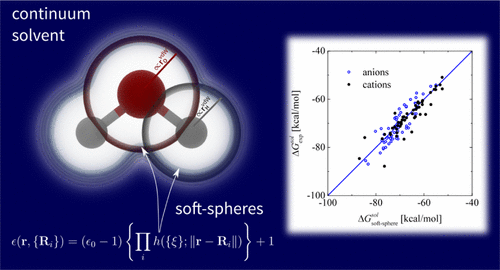
We present an implicit solvation approach where the interface between the quantum-mechanical solute and the surrounding environment is described by a fully continuous permittivity built up with atomic-centered “soft” spheres. This approach combines many of the advantages of the self-consistent continuum solvation model in handling solutes and surfaces in contact with complex dielectric environments or electrolytes in electronic-structure calculations. In addition it is able to describe accurately both neutral and charged systems. The continuous function, describing the variation of the permittivity, allows to compute analytically the nonelectrostatic contributions to the solvation free energy that are described in terms of the quantum surface. The whole methodology is computationally stable, provides consistent energies and forces, and keeps the computational efforts and runtimes comparable to those of standard vacuum calculations. The capabilitiy to treat arbitrary molecular or slab-like geometries as well as charged molecules is key to tackle electrolytes within mixed explicit/implicit frameworks. We show that, with given, fixed atomic radii, two parameters are sufficient to give a mean absolute error of only 1.12 kcal/mol with respect to the experimental aqueous solvation energies for a set of 274 neutral solutes. For charged systems, the same set of parameters provides solvation energies for a set of 60 anions and 52 cations with an error of 2.96 and 2.13 kcal/mol, respectively, improving upon previous literature values. To tackle elements not present in most solvation databases, a new benchmark scheme on wettability and contact angles is proposed for solid–liquid interfaces and applied to the investigation of the stable terminations of a CdS (112̅0) surface in an electrochemical medium.
中文翻译:

电子结构计算中的软球连续体解
我们提出了一种隐含的溶剂化方法,其中通过以原子为中心的“软”球体建立的完全连续的介电常数来描述量子力学溶质与周围环境之间的界面。这种方法结合了自洽连续介质模型的许多优点,可处理在电子结构计算中与复杂介电环境或电解质接触的溶质和表面。此外,它能够准确描述中性和带电系统。描述介电常数变化的连续函数可以分析地计算出对溶剂化自由能的非静电贡献,这些贡献以量子表面的形式描述。整个方法在计算上是稳定的,提供一致的能量和力,并使计算工作量和运行时间可与标准真空计算相媲美。处理任意分子或类似板的几何形状以及带电分子的能力是解决混合显式/隐式框架内电解质的关键。我们表明,在给定的固定原子半径的情况下,相对于一组274种中性溶质的实验水溶剂化能,两个参数足以给出平均绝对误差仅为1.12 kcal / mol。对于带电系统,同一组参数为一组60个阴离子和52个阳离子提供溶剂化能,误差分别为2.96和2.13 kcal / mol,比以前的文献值有所改善。为了解决大多数溶剂化数据库中不存在的元素,
更新日期:2017-07-03
中文翻译:
电子结构计算中的软球连续体解
我们提出了一种隐含的溶剂化方法,其中通过以原子为中心的“软”球体建立的完全连续的介电常数来描述量子力学溶质与周围环境之间的界面。这种方法结合了自洽连续介质模型的许多优点,可处理在电子结构计算中与复杂介电环境或电解质接触的溶质和表面。此外,它能够准确描述中性和带电系统。描述介电常数变化的连续函数可以分析地计算出对溶剂化自由能的非静电贡献,这些贡献以量子表面的形式描述。整个方法在计算上是稳定的,提供一致的能量和力,并使计算工作量和运行时间可与标准真空计算相媲美。处理任意分子或类似板的几何形状以及带电分子的能力是解决混合显式/隐式框架内电解质的关键。我们表明,在给定的固定原子半径的情况下,相对于一组274种中性溶质的实验水溶剂化能,两个参数足以给出平均绝对误差仅为1.12 kcal / mol。对于带电系统,同一组参数为一组60个阴离子和52个阳离子提供溶剂化能,误差分别为2.96和2.13 kcal / mol,比以前的文献值有所改善。为了解决大多数溶剂化数据库中不存在的元素,






































 京公网安备 11010802027423号
京公网安备 11010802027423号