当前位置:
X-MOL 学术
›
J. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Structure and Conformational Analysis of 1-Phenyl-1-X-1-Silacyclohexanes (X = F, Cl) by Electron Diffraction, Low-Temperature NMR, and Quantum Chemical Calculations
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2016-12-23 00:00:00 , DOI: 10.1021/acs.joc.6b02538 Bagrat A. Shainyan 1 , Alexander V. Belyakov 2 , Yurii F. Sigolaev 2 , Alexander N. Khramov 2 , Erich Kleinpeter 3
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2016-12-23 00:00:00 , DOI: 10.1021/acs.joc.6b02538 Bagrat A. Shainyan 1 , Alexander V. Belyakov 2 , Yurii F. Sigolaev 2 , Alexander N. Khramov 2 , Erich Kleinpeter 3
Affiliation
|
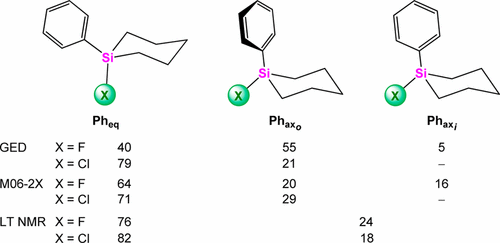
The molecular structure and conformational preferences of 1-phenyl-1-X-1-silacyclohexanes C5H10Si(Ph,X) (X = F (3), Cl (4)) were studied by gas-phase electron diffraction, low-temperature NMR spectroscopy, and high-level quantum chemical calculations. In the gas phase only three (3) and two (4) stable conformers differing in the axial or equatorial location of the phenyl group and the angle of rotation about the Si–CPh bond (axi and axo denote the Ph group lying in or out of the X–Si–CPh plane) contribute to the equilibrium. In 3 the ratio Pheq:Phaxo:Phaxi is 40(12):55(24):5 and 64:20:16 by experiment and theory, respectively. In 4 the ratio Pheq:Phaxo is 79(15):21(15) and 71:29 by experiment and theory (M06-2X calculations), respectively. The gas-phase electron diffraction parameters are in good agreement with those obtained from theory at the M06-2X/aug-ccPVTZ and MP2/aug-cc-pVTZ levels. Unlike the case for M06-2X, MP2 calculations indicate that 3-Pheq conformer lies 0.5 kcal/mol higher than the 3-Phaxo conformer. As follows from QTAIM analysis, the phenyl group is more stable when it is located in the axial position but produces destabilization of the silacyclohexane ring. By low-temperature NMR spectroscopy the six-membered-ring interconversion could be frozen at 103 K and the present conformational equilibria of 3 and 4 could be determined. The ratio of the conformers is 3-Pheq:3-Phax = (75–77):(23–25) and 4-Pheq:4-Phax = 82:18.
中文翻译:

通过电子衍射,低温NMR和量子化学计算对1-苯基-1-X-1-硅环己烷(X = F,Cl)进行分子结构和构象分析
通过气相电子衍射研究了1-苯基-1-X-1-硅环己烷C 5 H 10 Si(Ph,X)(X = F(3),Cl(4))的分子结构和构象偏好,低温NMR光谱和高级量子化学计算。在气相中,只有三(3)和两(4)个稳定构象异构体,其苯基基团的轴向或赤道位置以及围绕Si–C Ph键的旋转角(轴i和轴o表示位于基团上的Ph基团)在X–Si–C Ph平面之内或之外)有助于平衡。在3的比率Ph中eq:Ph ax o:Ph ax i通过实验和理论分别为40(12):55(24):5和64:20:16 在4中,根据实验和理论(M06-2X计算),Ph eq:Ph ax o的比率分别为79(15):21(15)和71:29。气相电子衍射参数与M06-2X / aug-ccPVTZ和MP2 / aug-cc-pVTZ水平的理论值相符。与M06-2X的情况不同,MP2计算表明3- Ph eq构象异构体比3- Ph ax o高0.5 kcal / mol 。适形者。如从QTAIM分析得出的,当苯基位于轴向位置时,它更稳定,但是会产生硅环己烷环的不稳定。通过低温NMR光谱,可以将六元环互变冻结在103K,并且可以确定目前的构象平衡为3和4。构象体的比例为3 -Ph eq:3 -Ph ax =(75–77):( 23–25)和4 -Ph eq:4 -Ph ax = 82:18。
更新日期:2016-12-23
中文翻译:
通过电子衍射,低温NMR和量子化学计算对1-苯基-1-X-1-硅环己烷(X = F,Cl)进行分子结构和构象分析
通过气相电子衍射研究了1-苯基-1-X-1-硅环己烷C 5 H 10 Si(Ph,X)(X = F(3),Cl(4))的分子结构和构象偏好,低温NMR光谱和高级量子化学计算。在气相中,只有三(3)和两(4)个稳定构象异构体,其苯基基团的轴向或赤道位置以及围绕Si–C Ph键的旋转角(轴i和轴o表示位于基团上的Ph基团)在X–Si–C Ph平面之内或之外)有助于平衡。在3的比率Ph中eq:Ph ax o:Ph ax i通过实验和理论分别为40(12):55(24):5和64:20:16 在4中,根据实验和理论(M06-2X计算),Ph eq:Ph ax o的比率分别为79(15):21(15)和71:29。气相电子衍射参数与M06-2X / aug-ccPVTZ和MP2 / aug-cc-pVTZ水平的理论值相符。与M06-2X的情况不同,MP2计算表明3- Ph eq构象异构体比3- Ph ax o高0.5 kcal / mol 。适形者。如从QTAIM分析得出的,当苯基位于轴向位置时,它更稳定,但是会产生硅环己烷环的不稳定。通过低温NMR光谱,可以将六元环互变冻结在103K,并且可以确定目前的构象平衡为3和4。构象体的比例为3 -Ph eq:3 -Ph ax =(75–77):( 23–25)和4 -Ph eq:4 -Ph ax = 82:18。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号