当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Nonadiabatic ab initio chemical reaction dynamics for the photoisomerization reaction of 3,5-dimethylisoxazole via the S1 electronic state
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-11-20 , DOI: 10.1039/d4cp03137g Mizuki Kimura, Shinkoh Nanbu
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-11-20 , DOI: 10.1039/d4cp03137g Mizuki Kimura, Shinkoh Nanbu
|
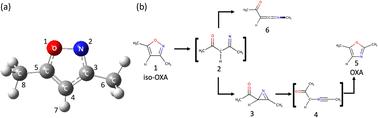
Nonadiabatic ab initio molecular dynamics simulations were performed to explore the photoisomerization pathway from isoxazole (iso-OXA) to oxazole (OXA), considering four electronic states. The XMS-CASPT2 and SA4-CASSCF theories were employed to describe these electronic structures, which were caused by 12 electrons in 11 orbitals with the cc-pVDZ + sp diffuse basis set; the Gaussian s- and p-type diffuse functions were extracted from Dunning's aug-cc-pVDZ function. The potential energy and its gradient at each time step were computed on-the-fly at these levels in the time evolution of the classical trajectory. When the two electronic states were close to each other, the trajectory surface hopping (TSH) judgment between the two adjacent states was carried out by the anteater procedure based on the Zhu–Nakamura formula (ZN-TSH). The two different excited state lifetimes were found to exist in the first electronic state (S1), estimated at 10.77 and 119.81 fs. Upon photoexcitation, the N–O bond breaks and energetically relaxes to the ground state (S0). In the pathway leading to the main product, azirine formation, the 5-membered ring retains a planar structure while undergoing a non-adiabatic transition with an increasing N–O bond distance. Furthermore, it was verified that a 1,2-shift takes place in the pathway that results in the production of ketenimine, causing a nonadiabatic transition.
中文翻译:

通过 S1 电子态对 3,5-二甲基异噁唑进行光异构化反应的非绝热从头化学反应动力学
考虑了四种电子状态,进行了非绝热从头分子动力学模拟,以探索从异恶唑 (iso-OXA) 到噁唑 (OXA) 的光异构化途径。采用 XMS-CASPT2 和 SA4-CASSCF 理论来描述这些电子结构,这些电子结构是由 cc-pVDZ + sp 漫射基集的 11 个轨道中的 12 个电子引起的;高斯 s 型和 p 型漫反射函数是从 Dunning 的 aug-cc-pVDZ 函数中提取的。在经典轨迹的时间演化中,每个时间步的势能及其梯度都是在这些水平上动态计算的。当两个电子态彼此接近时,通过基于 Zhu-Nakamura 公式 (ZN-TSH) 的食蚁兽程序进行两个相邻态之间的轨迹表面跳跃 (TSH) 判断。发现两种不同的激发态寿命存在于第一种电子态 (S1) 中,估计为 10.77 和 119.81 fs。光激发时,N-O 键断裂并在能量上弛豫至基态 (S0)。在通向主要产物氮杂嗪形成的途径中,5 元环在经历非绝热转变的同时保持平面结构,N-O 键距离增加。此外,经验证,在导致酮亚胺产生的途径中发生 1,2 次偏移,导致非绝热转变。
更新日期:2024-11-20
中文翻译:
通过 S1 电子态对 3,5-二甲基异噁唑进行光异构化反应的非绝热从头化学反应动力学
考虑了四种电子状态,进行了非绝热从头分子动力学模拟,以探索从异恶唑 (iso-OXA) 到噁唑 (OXA) 的光异构化途径。采用 XMS-CASPT2 和 SA4-CASSCF 理论来描述这些电子结构,这些电子结构是由 cc-pVDZ + sp 漫射基集的 11 个轨道中的 12 个电子引起的;高斯 s 型和 p 型漫反射函数是从 Dunning 的 aug-cc-pVDZ 函数中提取的。在经典轨迹的时间演化中,每个时间步的势能及其梯度都是在这些水平上动态计算的。当两个电子态彼此接近时,通过基于 Zhu-Nakamura 公式 (ZN-TSH) 的食蚁兽程序进行两个相邻态之间的轨迹表面跳跃 (TSH) 判断。发现两种不同的激发态寿命存在于第一种电子态 (S1) 中,估计为 10.77 和 119.81 fs。光激发时,N-O 键断裂并在能量上弛豫至基态 (S0)。在通向主要产物氮杂嗪形成的途径中,5 元环在经历非绝热转变的同时保持平面结构,N-O 键距离增加。此外,经验证,在导致酮亚胺产生的途径中发生 1,2 次偏移,导致非绝热转变。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号