npj Computational Materials ( IF 9.4 ) Pub Date : 2024-10-19 , DOI: 10.1038/s41524-024-01432-1 Hoje Chun, Jaclyn R. Lunger, Jeung Ku Kang, Rafael Gómez-Bombarelli, Byungchan Han
|
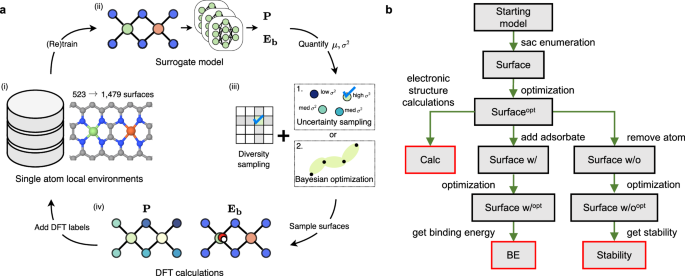
Single-atom catalysts (SACs) with multiple active sites exhibit high activity for a wide range of sluggish reactions, but identifying optimal multimetallic SAC is challenging due to the vast design space. Here, we present a self-driving computational strategy that combines first-principles calculations and equivariant graph neural network (GNN) to explore over 30,000 binary metallic sites with varying combinations of 3d transition metals and different ligand environments for oxygen reduction and evolution reactions (ORR/OER). Active learning facilitates the investigation of the search space by balancing the exploration of unseen atomic structures with the exploitation of the active ones. The GNN learns the chemical environments to capture composition-structure-property relationships for ORR/OER activity and selectivity. The computational predictions of promising Co-Fe, Co-Co, and Co-Zn metal pairs are consistent with the state-of-the-art results of experimental measurements reported in the literature. This approach can be extended to a broader class of multi-element high entropic materials systems.
中文翻译:
主动学习加速了对氧电催化多金属体系中单原子局部环境的探索
具有多个活性位点的单原子催化剂 (SAC) 对各种缓慢反应表现出高活性,但由于设计空间巨大,确定最佳多金属 SAC 具有挑战性。在这里,我们提出了一种自我驱动的计算策略,它结合了第一性原理计算和等变图神经网络 (GNN),以探索超过 30,000 个二元金属位点,这些位点具有不同的 3D 过渡金属组合和氧还原和析出反应 (ORR/OER) 的不同配体环境。主动学习通过平衡对看不见的原子结构的探索和对主动原子结构的利用来促进对搜索空间的研究。GNN 学习化学环境以捕获 ORR/OER 活性和选择性的成分-结构-性质关系。有前途的 Co-Fe、Co-Co 和 Co-Zn 金属对的计算预测与文献中报道的最新实验测量结果一致。这种方法可以扩展到更广泛的多单元高熵材料系统。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号