Nature Medicine ( IF 58.7 ) Pub Date : 2024-10-09 , DOI: 10.1038/s41591-024-03304-z Jerry R. Mendell, Francesco Muntoni, Craig M. McDonald, Eugenio M. Mercuri, Emma Ciafaloni, Hirofumi Komaki, Carmen Leon-Astudillo, Andrés Nascimento, Crystal Proud, Ulrike Schara-Schmidt, Aravindhan Veerapandiyan, Craig M. Zaidman, Maitea Guridi, Alexander P. Murphy, Carol Reid, Christoph Wandel, Damon R. Asher, Eddie Darton, Stefanie Mason, Rachael A. Potter, Teji Singh, Wenfei Zhang, Paulo Fontoura, Jacob S. Elkins, Louise R. Rodino-Klapac
|
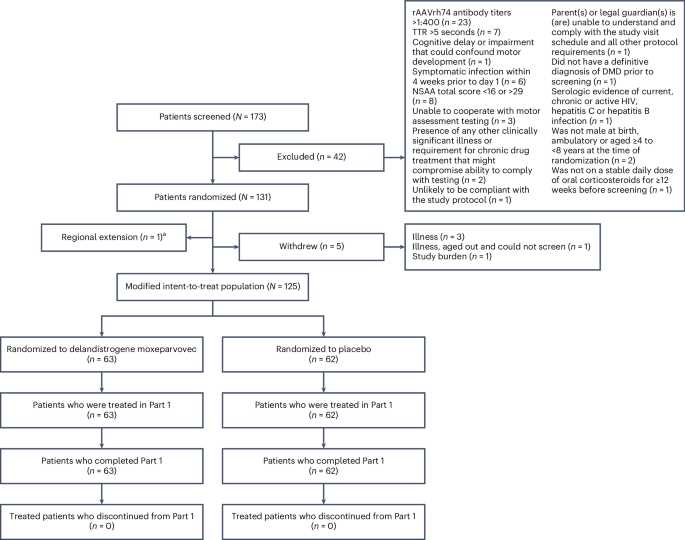
Duchenne muscular dystrophy (DMD) is a rare, X-linked neuromuscular disease caused by pathogenic variants in the DMD gene that result in the absence of functional dystrophin, beginning at birth and leading to progressive impaired motor function, loss of ambulation and life-threatening cardiorespiratory complications. Delandistrogene moxeparvovec, an adeno-associated rh74-viral vector-based gene therapy, addresses absent functional dystrophin in DMD. Here the phase 3 EMBARK study aimed to assess the efficacy and safety of delandistrogene moxeparvovec in patients with DMD. Ambulatory males with DMD, ≥4 years to <8 years of age, were randomized and stratified by age group and North Star Ambulatory Assessment (NSAA) score to single-administration intravenous delandistrogene moxeparvovec (1.33 × 1014 vector genomes per kilogram; n = 63) or placebo (n = 62). At week 52, the primary endpoint, change from baseline in NSAA score, was not met (least squares mean 2.57 (delandistrogene moxeparvovec) versus 1.92 (placebo) points; between-group difference, 0.65; 95% confidence interval (CI), −0.45, 1.74; P = 0.2441). Secondary efficacy endpoints included mean micro-dystrophin expression at week 12: 34.29% (treated) versus 0.00% (placebo). Other secondary efficacy endpoints at week 52 (between-group differences (95% CI)) included: Time to Rise (−0.64 (−1.06, −0.23)), 10-meter Walk/Run (−0.42 (−0.71, −0.13)), stride velocity 95th centile (0.10 (0.00, 0.19)), 100-meter Walk/Run (−3.29 (−8.28, 1.70)), time to ascend 4 steps (–0.36 (−0.71, −0.01)), PROMIS Mobility and Upper Extremity (0.05 (−0.08, 0.19); −0.04 (−0.24, 0.17)) and number of NSAA skills gained/improved (0.19 (−0.67, 1.06)). In total, 674 adverse events were recorded with delandistrogene moxeparvovec and 514 with placebo. There were no deaths, discontinuations or clinically significant complement-mediated adverse events; 7 patients (11.1%) experienced 10 treatment-related serious adverse events. Delandistrogene moxeparvovec did not lead to a significant improvement in NSAA score at week 52. Some of the secondary endpoints numerically favored treatment, although no statistical significance can be claimed. Safety was manageable and consistent with previous delandistrogene moxeparvovec trials. ClinicalTrials.gov: NCT05096221
中文翻译:
Duchenne 肌营养不良症的 AAV 基因治疗:EMBARK 3 期随机试验
杜氏肌营养不良症 (DMD) 是一种罕见的 X 连锁神经肌肉疾病,由 DMD 基因的致病性变异引起,导致功能性肌营养不良蛋白缺失,从出生开始,导致进行性运动功能受损、行走能力丧失和危及生命的心肺并发症。Delandistrogene moxeparvovec 是一种基于腺相关 rh74 病毒载体的基因疗法,可解决 DMD 中不存在的功能性肌萎缩蛋白。在这里,3 期 EMBARK 研究旨在评估 delandistrogene moxeparvovec 在 DMD 患者中的疗效和安全性。患有 DMD 的 ≥4 岁至 <8 岁的非卧床男性按年龄组和北极星动态评估 (NSAA) 评分随机和分层至单次静脉注射去蓝雌激素莫xeparvovec (1.33 ×10 14 个载体基因组/千克;n = 63)或安慰剂 (n = 62)。在第 52 周时,未达到主要终点,即 NSAA 评分相对于基线的变化(最小二乘均值 2.57 (delandistrogene moxeparvovec) vs 1.92 (安慰剂) 分;组间差异,0.65;95% 置信区间 (CI),-0.45,1.74;P = 0.2441)。次要疗效终点包括第 12 周时的平均微量抗肌萎缩蛋白表达:34.29%(治疗组)对 0.00%(安慰剂组)。第 52 周时的其他次要疗效终点(组间差异 (95% CI))包括:起床时间 (-0.64 (-1.06, -0.23))、10 米步行/跑步 (-0.42 (-0.71, -0.13))、步速第 95 个百分位 (0.10 (0.00, 0.19))、100 米步行/跑步 (-3.29 (-8.28, 1.70))、上升 4 级时间 (-0.36 (-0.71, -0.01))、PROMIS 活动性和上肢 (0.05 (-0.08, 0.19);-0.04 (-0.24, 0.17)) 和获得/改进的 NSAA 技能数量 (0.19 (-0.67, 1.06))。 总共记录了 674 例不良事件,delandistrogene moxeparvovec 组记录了 514 例不良事件。没有死亡、停药或有临床意义的补体介导的不良事件;7 例患者 (11.1%) 经历了 10 例与治疗相关的严重不良事件。Delandistrogene moxeparvovec 在第 52 周时未导致 NSAA 评分的显着改善。一些次要终点在数值上有利于治疗,尽管不能声称具有统计学意义。安全性可控,与之前的 delandistrogene moxeparvovec 试验一致。临床试验。政府: NCT05096221

















































 京公网安备 11010802027423号
京公网安备 11010802027423号