Leukemia ( IF 12.8 ) Pub Date : 2024-09-17 , DOI: 10.1038/s41375-024-02405-5 Melisa Halilovic, Mohamed Abdelsalam, Joanna Zabkiewicz, Michelle Lazenby, Caroline Alvares, Matthias Schmidt, Walburgis Brenner, Sara Najafi, Ina Oehme, Christoph Hieber, Yanira Zeyn, Matthias Bros, Wolfgang Sippl, Oliver H. Krämer
|
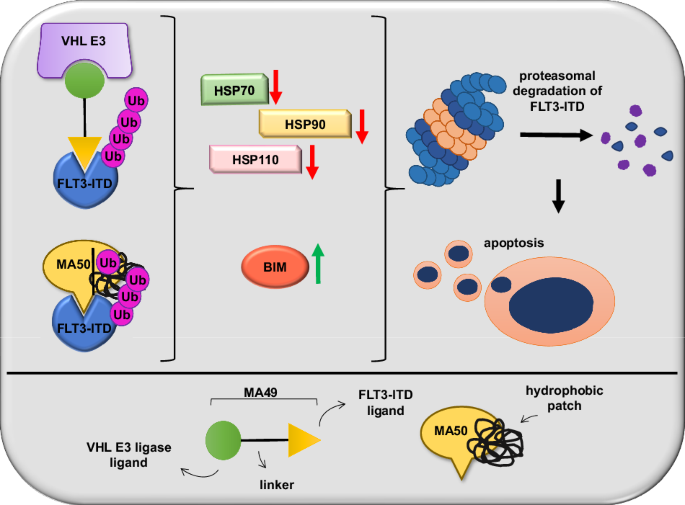
Internal tandem duplications in the FMS-like tyrosine kinase-3 (FLT3-ITD) are common mutations in acute myeloid leukemia (AML). Proteolysis-targeting chimeras (PROTACs) that induce proteasomal degradation of mutated FLT3 emerge as innovative pharmacological approach. Molecular mechanisms that control targeted proteolysis beyond the ubiquitin-proteasome-system are undefined and PROTACs are the only known type of FLT3 degraders. We report that the von-Hippel-Lindau ubiquitin-ligase based FLT3 PROTAC MA49 (melotinib-49) and the FLT3 hydrophobic tagging molecule MA50 (halotinib-50) reduce endoplasmic reticulum-associated, oncogenic FLT3-ITD but spare FLT3. Nanomolar doses of MA49 and MA50 induce apoptosis of human leukemic cell lines and primary AML blasts with FLT3-ITD (p < 0.05-0.0001), but not of primary hematopoietic stem cells and differentiated immune cells, FLT3 wild-type cells, retinal cells, and c-KIT-dependent cells. In vivo activity of MA49 against FLT3-ITD-positive leukemia cells is verified in a Danio rerio model. The degrader-induced loss of FLT3-ITD involves the pro-apoptotic BH3-only protein BIM and a previously unidentified degrader-induced depletion of protein-folding chaperones. The expression levels of HSP90 and HSP110 correlate with reduced AML patient survival (p < 0.1) and HSP90, HSP110, and BIM are linked to the expression of FLT3 in primary AML cells (p < 0.01). HSP90 suppresses degrader-induced FLT3-ITD elimination and thereby establishes a mechanistically defined feed-back circuit.
中文翻译:
突变型 FMS 样酪氨酸激酶 3 的选择性降解需要 BIM 依赖性的热休克蛋白消耗
FMS 样酪氨酸激酶 3 (FLT3-ITD) 的内部串联重复是急性髓系白血病 (AML) 的常见突变。诱导突变 FLT3 蛋白酶体降解的蛋白水解靶向嵌合体 (PROTAC) 成为创新的药理学方法。控制泛素蛋白酶体系统之外的靶向蛋白水解的分子机制尚不清楚,PROTAC 是唯一已知的 FLT3 降解剂类型。我们报道基于 von-Hippel-Lindau 泛素连接酶的 FLT3 PROTAC MA49 (melotinib-49) 和 FLT3 疏水标记分子 MA50 (halotinib-50) 减少内质网相关的致癌性 FLT3-ITD,但保留 FLT3。纳摩尔剂量的 MA49 和 MA50 使用 FLT3-ITD 诱导人白血病细胞系和原代 AML 母细胞凋亡 ( p < 0.05-0.0001),但不诱导原代造血干细胞和分化免疫细胞、FLT3 野生型细胞、视网膜细胞凋亡和 c-KIT 依赖性细胞。 MA49 针对 FLT3-ITD 阳性白血病细胞的体内活性在斑马鱼模型中得到验证。降解剂诱导的 FLT3-ITD 丢失涉及促凋亡 BH3 蛋白 BIM 和先前未识别的降解剂诱导的蛋白质折叠伴侣的损耗。 HSP90 和 HSP110 的表达水平与 AML 患者生存率降低相关 ( p < 0.1),HSP90、HSP110 和 BIM 与原代 AML 细胞中 FLT3 的表达相关 ( p < 0.01)。 HSP90 抑制降解剂引起的 FLT3-ITD 消除,从而建立机械定义的反馈电路。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号