npj Computational Materials ( IF 9.4 ) Pub Date : 2024-09-08 , DOI: 10.1038/s41524-024-01391-7 Dongsheng Wen , Victoria Tucker , Michael S. Titus
|
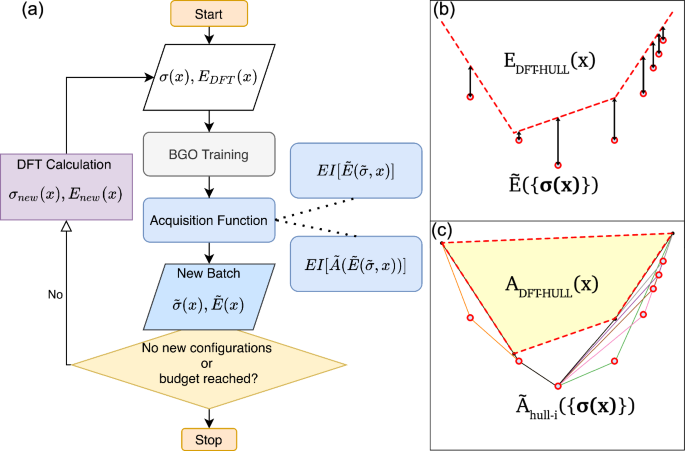
Atomistic simulations are crucial for predicting material properties and understanding phase stability, essential for materials selection and development. However, the high computational cost of density functional theory calculations challenges the design of materials with complex structures and composition. This study introduces new data acquisition strategies using Bayesian-Gaussian optimization that efficiently integrate the geometry of the convex hull to optimize the yield of batch experiments. We developed uncertainty-based acquisition functions to prioritize the computation tasks of configurations of multi-component alloys, enhancing our ability to identify the ground-state line. Our methods were validated across diverse materials systems including Co-Ni alloys, Zr-O compounds, Ni-Al-Cr ternary alloys, and a planar defect system in intermetallic (Ni1−x, Cox)3Al. Compared to traditional genetic algorithms, our strategies reduce training parameters and user interaction, cutting the number of experiments needed to accurately determine the ground-state line by over 30%. These approaches can be expanded to multi-component systems and integrated with cost functions to further optimize experimental designs.
中文翻译:
多组分合金簇扩展凸包加速搜索的贝叶斯优化获取函数
原子模拟对于预测材料特性和了解相稳定性至关重要,这对于材料选择和开发至关重要。然而,密度泛函理论计算的高计算成本对具有复杂结构和成分的材料的设计提出了挑战。本研究引入了使用贝叶斯-高斯优化的新数据采集策略,该策略有效地集成凸包的几何形状以优化批量实验的产量。我们开发了基于不确定性的采集函数来优先考虑多组分合金配置的计算任务,从而增强我们识别基态线的能力。我们的方法在不同的材料系统中得到了验证,包括 Co-Ni 合金、Zr-O 化合物、Ni-Al-Cr 三元合金以及金属间化合物 (Ni 1− x , Co x ) 3 Al 中的平面缺陷系统。与传统遗传算法相比,我们的策略减少了训练参数和用户交互,将准确确定基态线所需的实验数量减少了 30% 以上。这些方法可以扩展到多组件系统,并与成本函数集成,以进一步优化实验设计。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号