当前位置:
X-MOL 学术
›
Energy Fuels
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Aggregation of Asphaltene Subfractions A1 and A2 in Different Solvents from the Perspective of Molecular Dynamics Simulations
Energy & Fuels ( IF 5.2 ) Pub Date : 2023-02-02 , DOI: 10.1021/acs.energyfuels.2c03054 Orlando Villegas 1, 2 , Germain Salvato Vallverdu 2, 3 , Brice Bouyssiere 2, 3 , Sócrates Acevedo 1 , Jimmy Castillo 1 , Isabelle Baraille 2, 3
Energy & Fuels ( IF 5.2 ) Pub Date : 2023-02-02 , DOI: 10.1021/acs.energyfuels.2c03054 Orlando Villegas 1, 2 , Germain Salvato Vallverdu 2, 3 , Brice Bouyssiere 2, 3 , Sócrates Acevedo 1 , Jimmy Castillo 1 , Isabelle Baraille 2, 3
Affiliation
|
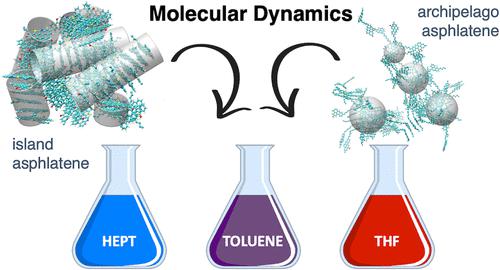
The aggregation behavior of model molecules of asphaltene subfractions A1 and A2 dissolved in heptane, toluene, and tetrahydrofuran (THF) were studied using molecular dynamics simulations. The proposed asphaltene molecular models are based on previously studied structures with two new models, including a highly aromatic model with a prominent island-type molecule and another molecule with a prominent archipelago-type architecture. The aggregation mechanisms in toluene, THF, and heptane solvents were studied. The results in heptane and toluene were consistent with the solubility of asphaltenes and their subfractions in these solvents. The size of the aggregates is well-correlated with aromaticity. When considering THF, large aggregates are broken down into smaller aggregates. This could lead to the mixture of high, medium, and low molecular weight distribution bands usually observed when gel permeation chromatography (GPC) analyses are conducted on asphaltene samples in THF. The size distribution extracted from the simulations shows a bimodal distribution with profiles similar to the size distribution profiles usually found in GPC analysis for asphaltene samples in THF. The distributions of dipole moments of the aggregates against the number of molecules in aggregates were constructed in both THF and toluene and reveal that the dipole moment of the aggregates vanishes when the number of molecules increases as a result of a random structure of the aggregates. The contributions of different molecular interactions to the aggregation mechanism, such as π stacking, van der Waals, and hydrogen bonds, are described.
中文翻译:

从分子动力学模拟的角度看沥青质亚组分 A1 和 A2 在不同溶剂中的聚集
使用分子动力学模拟研究了溶解在庚烷、甲苯和四氢呋喃 (THF) 中的沥青质亚组分 A1 和 A2 模型分子的聚集行为。拟议的沥青质分子模型基于先前研究的结构和两个新模型,包括具有突出岛型分子的高度芳香模型和具有突出群岛型结构的另一个分子。研究了甲苯、四氢呋喃和庚烷溶剂中的聚集机制。在庚烷和甲苯中的结果与沥青质及其亚馏分在这些溶剂中的溶解度一致。聚集体的大小与芳香性密切相关。在考虑 THF 时,大的聚集体被分解成更小的聚集体。这可能会导致高、中、在 THF 中对沥青质样品进行凝胶渗透色谱 (GPC) 分析时,通常会观察到低分子量分布带。从模拟中提取的粒径分布显示双峰分布,其轮廓类似于在 THF 中的沥青质样品的 GPC 分析中通常发现的粒径分布轮廓。在 THF 和甲苯中构建了聚集体偶极矩相对于聚集体中分子数的分布,表明当分子数由于聚集体的随机结构而增加时,聚集体的偶极矩消失。描述了不同分子相互作用对聚集机制的贡献,例如 π 堆积、范德华力和氢键。
更新日期:2023-02-02
中文翻译:
从分子动力学模拟的角度看沥青质亚组分 A1 和 A2 在不同溶剂中的聚集
使用分子动力学模拟研究了溶解在庚烷、甲苯和四氢呋喃 (THF) 中的沥青质亚组分 A1 和 A2 模型分子的聚集行为。拟议的沥青质分子模型基于先前研究的结构和两个新模型,包括具有突出岛型分子的高度芳香模型和具有突出群岛型结构的另一个分子。研究了甲苯、四氢呋喃和庚烷溶剂中的聚集机制。在庚烷和甲苯中的结果与沥青质及其亚馏分在这些溶剂中的溶解度一致。聚集体的大小与芳香性密切相关。在考虑 THF 时,大的聚集体被分解成更小的聚集体。这可能会导致高、中、在 THF 中对沥青质样品进行凝胶渗透色谱 (GPC) 分析时,通常会观察到低分子量分布带。从模拟中提取的粒径分布显示双峰分布,其轮廓类似于在 THF 中的沥青质样品的 GPC 分析中通常发现的粒径分布轮廓。在 THF 和甲苯中构建了聚集体偶极矩相对于聚集体中分子数的分布,表明当分子数由于聚集体的随机结构而增加时,聚集体的偶极矩消失。描述了不同分子相互作用对聚集机制的贡献,例如 π 堆积、范德华力和氢键。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号