Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2022-05-07 , DOI: 10.1016/j.comptc.2022.113735 Yalan Liu 1 , Zhihao Zhong 1 , Shiqi Xu 1
|
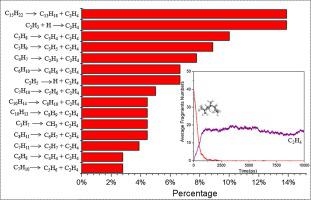
Tetrahydrotricyclopentadiene (THTCPD) has high density and volumetric heat, but its viscosity is also high, resisting its ignition and combustion, which somewhat limits its applications. Therefore, it is crucial to understand the mechanism of THTCPD pyrolysis. In this study, the ReaxFF reactive molecular dynamics simulations were performed to investigate the thermal dissociation of THTCPD. The detailed description of THTCPD pyrolysis behavior was obtained from the aspects of initial reactions, main species distributions and intermolecular reactions. Initiation of the THTCPD decomposition is mainly through C C bond fission of the bicycloheptyl structure in the middle, leading to the formation of highly active C15H22 diradicals. To give a better description of the pyrolysis behavior, main products related intermolecular reactions should be considered in the mechanism. The simulated pre-exponential factor A (2.30 × 1015 s−1) and activation energy E (55.72 kcal/mol) captured from simulations are found to be reasonably consistent with the experimental values.
C bond fission of the bicycloheptyl structure in the middle, leading to the formation of highly active C15H22 diradicals. To give a better description of the pyrolysis behavior, main products related intermolecular reactions should be considered in the mechanism. The simulated pre-exponential factor A (2.30 × 1015 s−1) and activation energy E (55.72 kcal/mol) captured from simulations are found to be reasonably consistent with the experimental values.
中文翻译:
ReaxFF反应分子动力学模拟四氢三环戊二烯的热解机理
四氢三环戊二烯(THTCPD)具有高密度和体积热,但其粘度也很高,难以点燃和燃烧,这在一定程度上限制了它的应用。因此,了解 THTCPD 热解的机制至关重要。在本研究中,进行了 ReaxFF 反应分子动力学模拟以研究 THTCPD 的热解离。从初始反应、主要物种分布和分子间反应等方面对THPCD热解行为进行了详细描述。THTCPD分解的引发主要是通过中间的双环庚基结构的C C键裂变形成高活性C 15 H 22激进分子。为了更好地描述热解行为,在机理中应考虑与分子间反应相关的主要产物。发现从模拟中捕获的模拟的指前因子 A (2.30 × 10 15 s -1 ) 和活化能 E (55.72 kcal/mol) 与实验值相当一致。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号