当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Importance of Ligand Reorganization Free Energy in Protein−Ligand Binding-Affinity Prediction
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2009-09-30 , DOI: 10.1021/ja9039373 Chao-Yie Yang 1 , Haiying Sun , Jianyong Chen , Zaneta Nikolovska-Coleska , Shaomeng Wang
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2009-09-30 , DOI: 10.1021/ja9039373 Chao-Yie Yang 1 , Haiying Sun , Jianyong Chen , Zaneta Nikolovska-Coleska , Shaomeng Wang
Affiliation
|
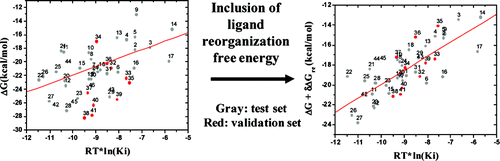
Accurate prediction of the binding affinities of small-molecule ligands to their biological targets is fundamental for structure-based drug design but remains a very challenging task. In this paper, we have performed computational studies to predict the binding models of 31 small-molecule Smac (the second mitochondria-derived activator of caspase) mimetics to their target, the XIAP (X-linked inhibitor of apoptosis) protein, and their binding affinities. Our results showed that computational docking was able to reliably predict the binding models, as confirmed by experimentally determined crystal structures of some Smac mimetics complexed with XIAP. However, all the computational methods we have tested, including an empirical scoring function, two knowledge-based scoring functions, and MM-GBSA (molecular mechanics and generalized Born surface area), yield poor to modest prediction for binding affinities. The linear correlation coefficient (r(2)) value between the predicted affinities and the experimentally determined affinities was found to be between 0.21 and 0.36. Inclusion of ensemble protein-ligand conformations obtained from molecular dynamic simulations did not significantly improve the prediction. However, major improvement was achieved when the free-energy change for ligands between their free- and bound-states, or "ligand-reorganization free energy", was included in the MM-GBSA calculation, and the r(2) value increased from 0.36 to 0.66. The prediction was validated using 10 additional Smac mimetics designed and evaluated by an independent group. This study demonstrates that ligand reorganization free energy plays an important role in the overall binding free energy between Smac mimetics and XIAP. This term should be evaluated for other ligand-protein systems and included in the development of new scoring functions. To our best knowledge, this is the first computational study to demonstrate the importance of ligand reorganization free energy for the prediction of protein-ligand binding free energy.
中文翻译:

配体重组自由能在蛋白质-配体结合-亲和力预测中的重要性
准确预测小分子配体与其生物靶标的结合亲和力是基于结构的药物设计的基础,但仍然是一项非常具有挑战性的任务。在本文中,我们进行了计算研究来预测 31 种小分子 Smac(第二个线粒体衍生的半胱天冬酶激活剂)模拟物与其靶标 XIAP(X 连锁细胞凋亡抑制剂)蛋白的结合模型,以及它们的结合亲和力。我们的结果表明,计算对接能够可靠地预测结合模型,正如实验确定的与 XIAP 复合的一些 Smac 模拟物的晶体结构所证实的那样。然而,我们测试过的所有计算方法,包括一个经验评分函数、两个基于知识的评分函数和 MM-GBSA(分子力学和广义玻恩表面积),对结合亲和力产生较差或适度的预测。发现预测的亲和力和实验确定的亲和力之间的线性相关系数 (r(2)) 值介于 0.21 和 0.36 之间。包含从分子动力学模拟获得的整体蛋白质配体构象并没有显着改善预测。然而,当 MM-GBSA 计算中包含配体在其自由和结合状态之间的自由能变化或“配体重组自由能”时,实现了重大改进,并且 r(2) 值从0.36 到 0.66。使用由独立小组设计和评估的 10 个额外的 Smac 模拟物验证了该预测。该研究表明配体重组自由能在 Smac 模拟物和 XIAP 之间的整体结合自由能中起着重要作用。该术语应针对其他配体-蛋白质系统进行评估,并包括在新评分功能的开发中。据我们所知,这是第一个证明配体重组自由能对预测蛋白质-配体结合自由能的重要性的计算研究。
更新日期:2009-09-30
中文翻译:
配体重组自由能在蛋白质-配体结合-亲和力预测中的重要性
准确预测小分子配体与其生物靶标的结合亲和力是基于结构的药物设计的基础,但仍然是一项非常具有挑战性的任务。在本文中,我们进行了计算研究来预测 31 种小分子 Smac(第二个线粒体衍生的半胱天冬酶激活剂)模拟物与其靶标 XIAP(X 连锁细胞凋亡抑制剂)蛋白的结合模型,以及它们的结合亲和力。我们的结果表明,计算对接能够可靠地预测结合模型,正如实验确定的与 XIAP 复合的一些 Smac 模拟物的晶体结构所证实的那样。然而,我们测试过的所有计算方法,包括一个经验评分函数、两个基于知识的评分函数和 MM-GBSA(分子力学和广义玻恩表面积),对结合亲和力产生较差或适度的预测。发现预测的亲和力和实验确定的亲和力之间的线性相关系数 (r(2)) 值介于 0.21 和 0.36 之间。包含从分子动力学模拟获得的整体蛋白质配体构象并没有显着改善预测。然而,当 MM-GBSA 计算中包含配体在其自由和结合状态之间的自由能变化或“配体重组自由能”时,实现了重大改进,并且 r(2) 值从0.36 到 0.66。使用由独立小组设计和评估的 10 个额外的 Smac 模拟物验证了该预测。该研究表明配体重组自由能在 Smac 模拟物和 XIAP 之间的整体结合自由能中起着重要作用。该术语应针对其他配体-蛋白质系统进行评估,并包括在新评分功能的开发中。据我们所知,这是第一个证明配体重组自由能对预测蛋白质-配体结合自由能的重要性的计算研究。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号