当前位置:
X-MOL 学术
›
J. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Acceleration of saddle-point searches with machine learning
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2016-08-16 12:24:18 , DOI: 10.1063/1.4960708 Andrew A. Peterson 1
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2016-08-16 12:24:18 , DOI: 10.1063/1.4960708 Andrew A. Peterson 1
Affiliation
|
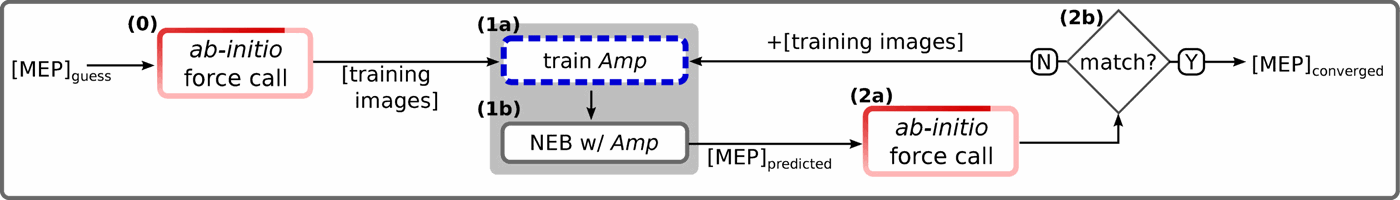
In atomistic simulations, the location of the saddle point on the potential-energy surface (PES) gives important information on transitions between local minima, for example, via transition-state theory. However, the search for saddle points often involves hundreds or thousands of ab initio force calls, which are typically all done at full accuracy. This results in the vast majority of the computational effort being spent calculating the electronic structure of states not important to the researcher, and very little time performing the calculation of the saddle point state itself. In this work, we describe how machine learning (ML) can reduce the number of intermediate ab initio calculations needed to locate saddle points. Since machine-learning models can learn from, and thus mimic, atomistic simulations, the saddle-point search can be conducted rapidly in the machine-learning representation. The saddle-point prediction can then be verified by an ab initio calculation; if it is incorrect, this strategically has identified regions of the PES where the machine-learning representation has insufficient training data. When these training data are used to improve the machine-learning model, the estimates greatly improve. This approach can be systematized, and in two simple example problems we demonstrate a dramatic reduction in the number of ab initio force calls. We expect that this approach and future refinements will greatly accelerate searches for saddle points, as well as other searches on the potential energy surface, as machine-learning methods see greater adoption by the atomistics community.
中文翻译:

利用机器学习加速鞍点搜索
在原子模拟中,鞍点在势能表面(PES)上的位置提供了有关局部最小值之间的过渡的重要信息,例如,通过过渡状态理论。但是,寻找鞍点通常涉及成百上千的从头算起强制调用,通常都可以完全准确地完成。这导致绝大多数计算工作都花在了计算对研究人员不重要的状态的电子结构上,而花费很少的时间进行鞍点状态本身的计算。在这项工作中,我们描述了机器学习(ML)如何减少定位鞍点所需的中间从头计算的数量。由于机器学习模型可以从原子模拟中学习,从而进行模仿,因此可以在机器学习表示中快速进行鞍点搜索。然后可以通过从头算计算来验证鞍点预测。如果不正确,则从战略上确定了机器学习表示的训练数据不足的PES区域。当使用这些训练数据来改进机器学习模型时,估计会大大提高。这种方法可以系统化,在两个简单的示例问题中,我们证明了显着减少了从头算起。我们希望这种方法和未来的改进将极大地加快对鞍点的搜索以及对势能面的其他搜索,因为机器学习方法已被原子学界广泛采用。
更新日期:2016-08-17
中文翻译:
利用机器学习加速鞍点搜索
在原子模拟中,鞍点在势能表面(PES)上的位置提供了有关局部最小值之间的过渡的重要信息,例如,通过过渡状态理论。但是,寻找鞍点通常涉及成百上千的从头算起强制调用,通常都可以完全准确地完成。这导致绝大多数计算工作都花在了计算对研究人员不重要的状态的电子结构上,而花费很少的时间进行鞍点状态本身的计算。在这项工作中,我们描述了机器学习(ML)如何减少定位鞍点所需的中间从头计算的数量。由于机器学习模型可以从原子模拟中学习,从而进行模仿,因此可以在机器学习表示中快速进行鞍点搜索。然后可以通过从头算计算来验证鞍点预测。如果不正确,则从战略上确定了机器学习表示的训练数据不足的PES区域。当使用这些训练数据来改进机器学习模型时,估计会大大提高。这种方法可以系统化,在两个简单的示例问题中,我们证明了显着减少了从头算起。我们希望这种方法和未来的改进将极大地加快对鞍点的搜索以及对势能面的其他搜索,因为机器学习方法已被原子学界广泛采用。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号