当前位置:
X-MOL 学术
›
Organometallics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Understanding Catalyst Structure–Selectivity Relationships in Pd-Catalyzed Enantioselective Methoxycarbonylation of Styrene
Organometallics ( IF 2.5 ) Pub Date : 2020-12-14 , DOI: 10.1021/acs.organomet.0c00613 Simone Gallarati 1 , Paul Dingwall 2 , José A. Fuentes 1 , Michael Bühl 1 , Matthew L. Clarke 1
Organometallics ( IF 2.5 ) Pub Date : 2020-12-14 , DOI: 10.1021/acs.organomet.0c00613 Simone Gallarati 1 , Paul Dingwall 2 , José A. Fuentes 1 , Michael Bühl 1 , Matthew L. Clarke 1
Affiliation
|
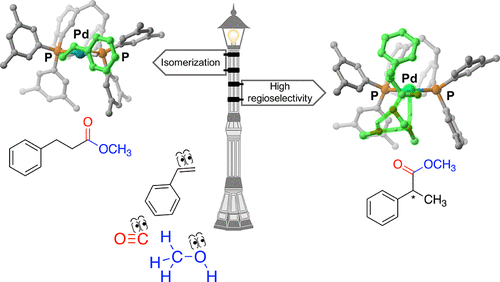
Catalyst-controlled regioselectivity in palladium-catalyzed carbonylation of alkenes has been a long-standing goal of homogeneous catalysis. In general, monophosphines do favor branched regioselectivity, but lead to poor enantioselectivity, while diphosphines give mainly linear products. Previously, we reported the simultaneous control of regio- and enantioselectivity in the hydroxy- and methoxycarbonylation of vinyl arenes with Pd complexes of the Phanephos ligand. Herein, we present a density functional theory study (B3PW91-D3 level of theory) of the catalytic cycle, supported by deuterium labeling studies, to understand its mechanism. Alkene coordination to a Pd-hydride species was identified as the origin of asymmetric induction and regioselectivity in both the parent Pd/Xylyl-Phanephos catalyst and electron-deficient analogue, and rationalized according to a quadrant-diagram representation. The mechanism by which the preferentially formed pro-(S) Pd-alkene complex can isomerize via rotation around the palladium–(C═C) bond was investigated. In the parent system, this process is in competition with the methanolysis step that leads to the ester product and is responsible for the overall loss of regioselectivity. On the other hand, the introduction of electron-withdrawing substituents on the catalyst framework results in the reduction of the methanolysis barriers, making the isomerization pathway energetically unfavorable and so leading simultaneously to high regiocontrol and good enantiomeric ratios.
中文翻译:

了解Pd催化的苯乙烯对映选择性甲氧羰基化反应中的催化剂结构-选择性关系
钯催化的烯烃羰基化反应中催化剂控制的区域选择性一直是均相催化的长期目标。通常,单膦酸酯确实有利于支链区域选择性,但是导致差的对映选择性,而二膦酸酯主要给出线性产物。以前,我们报道了同时控制Phanephos配体的Pd配合物在乙烯基芳烃的羟基和甲氧基羰基化中的区域选择性和对映选择性。在这里,我们提出了催化循环的密度泛函理论研究(B3PW91-D3的理论水平),并辅以氘标记研究,以了解其机理。烯烃与Pd氢化物的配位被确定为母体Pd / Xylyl-Phanephos催化剂和电子不足的类似物的不对称诱导和区域选择性的起源,并根据象限图表示进行合理化。优先形成pro-(S)研究了钯-烯烃配合物可以通过绕钯-(C═C)键旋转而异构化。在母体系统中,该过程与导致酯产物的甲醇分解步骤竞争,这是区域选择性总体损失的原因。另一方面,在催化剂骨架上引入吸电子取代基导致甲醇分解障碍的减少,使得异构化途径在能量上不利,因此同时导致高的区域控制和良好的对映体比率。
更新日期:2020-12-28
中文翻译:
了解Pd催化的苯乙烯对映选择性甲氧羰基化反应中的催化剂结构-选择性关系
钯催化的烯烃羰基化反应中催化剂控制的区域选择性一直是均相催化的长期目标。通常,单膦酸酯确实有利于支链区域选择性,但是导致差的对映选择性,而二膦酸酯主要给出线性产物。以前,我们报道了同时控制Phanephos配体的Pd配合物在乙烯基芳烃的羟基和甲氧基羰基化中的区域选择性和对映选择性。在这里,我们提出了催化循环的密度泛函理论研究(B3PW91-D3的理论水平),并辅以氘标记研究,以了解其机理。烯烃与Pd氢化物的配位被确定为母体Pd / Xylyl-Phanephos催化剂和电子不足的类似物的不对称诱导和区域选择性的起源,并根据象限图表示进行合理化。优先形成pro-(S)研究了钯-烯烃配合物可以通过绕钯-(C═C)键旋转而异构化。在母体系统中,该过程与导致酯产物的甲醇分解步骤竞争,这是区域选择性总体损失的原因。另一方面,在催化剂骨架上引入吸电子取代基导致甲醇分解障碍的减少,使得异构化途径在能量上不利,因此同时导致高的区域控制和良好的对映体比率。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号