当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Thermal Stability of Bis-Tetrazole and Bis-Triazole Derivatives with Long Catenated Nitrogen Chains: Quantitative Insights from High-Level Quantum Chemical Calculations.
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-08-11 , DOI: 10.1021/acs.jpca.0c04985 Margarita V Gorn 1, 2 , Nina P Gritsan 1, 2 , C Franklin Goldsmith 3 , Vitaly G Kiselev 1, 2, 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-08-11 , DOI: 10.1021/acs.jpca.0c04985 Margarita V Gorn 1, 2 , Nina P Gritsan 1, 2 , C Franklin Goldsmith 3 , Vitaly G Kiselev 1, 2, 3
Affiliation
|
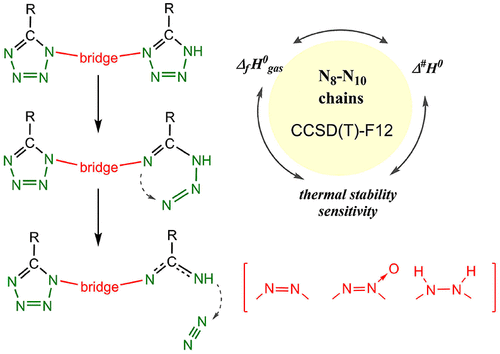
Azobis tetrazole and triazole derivatives containing long catenated nitrogen atom chains are of great interest as promising green energetic materials. However, these compounds often exhibit poor thermal stability and high impact sensitivity. Kinetics and mechanism of the primary decomposition reactions are directly related to these issues. In the present work, with the aid of highly accurate CCSD(T)-F12 quantum chemical calculations, we obtained reliable bond dissociation energies and activation barriers of thermolysis reactions for a number of N-rich heterocycles. We studied all existing 1,1′-azobistetrazoles containing an N10 chain, their counterparts with the 5,5′-bridging pattern, and the species with hydrazo- and azoxy-bridges, which are often present energetic moieties. The N8-containing azobistriazole was considered as well. For all compounds studied, the radical decomposition channel was found to be kinetically unfavorable. All species decompose via the ring-opening reaction yielding a transient azide (or diazo) intermediate followed by the N2 elimination. In the case of azobistetrazole derivatives, the calculated effective activation barriers of decomposition are ∼26–33 kcal mol–1, which is notably lower than that of tetrazole (∼40 kcal mol–1). This fact agrees well with the low thermal stability and high impact sensitivities of the former species. The activation barriers of the N2 elimination were found to be almost the same for the azobis compounds and the parent tetrazole, and the effective decomposition barrier is determined by the thermodynamics of the tetrazole–azide rearrangement. In comparison with 1,1′-azobistetrazole, the hydrazo-bridged compound is more stable kinetically due to the lack of pi-conjugation in the azide intermediate. In turn, the azoxy-bridged compounds are entirely unstable due to tremendous azide stabilization by the hydrogen bond formation. In general, the 5,5′-bridged species are more thermally stable than their 1,1′-counterparts due to a much higher barrier of the N2 elimination. Apart from this, the highly accurate gas-phase formation enthalpies were calculated at the W1–F12 level of theory for all species studied.
中文翻译:

带有长链氮链的Bis-Tetrazole和Bis-Triazole衍生物的热稳定性:高级量子化学计算的定量见解。
含有长链氮原子链的偶氮四唑和三唑衍生物作为有前途的绿色高能材料备受关注。但是,这些化合物通常表现出差的热稳定性和高冲击敏感性。初级分解反应的动力学和机理与这些问题直接相关。在当前的工作中,借助于高精度的CCSD(T)-F12量子化学计算,我们获得了许多N富集杂环的可靠键解离能和热解反应的激活势垒。我们研究了所有现有的含有N 10链的1,1'-偶氮双四唑,具有5,5'-桥接模式的对应物,以及带有azo-和乙氧基-桥的物种,它们通常是高能部分。N 8还考虑到含-偶氮双三唑。对于所有研究的化合物,发现自由基分解通道在动力学上是不利的。所有种类均通过开环反应分解,产生瞬态叠氮化物(或重氮)中间体,然后消除N 2。在偶氮双四唑衍生物的情况下,计算出的有效分解活化势垒为〜26–33 kcal mol –1,显着低于四唑(〜40 kcal mol –1)。这一事实与前一个物种的低热稳定性和高冲击敏感性非常吻合。N 2的激活障碍偶氮二化合物和母体四唑的消除几乎相同,有效的分解势垒由四唑-叠氮化物重排的热力学决定。与1,1'-偶氮双四唑相比,由于叠氮化物中间体缺乏π共轭,因此肼桥联的化合物在动力学上更稳定。继而,由于通过氢键形成的巨大的叠氮化物稳定性,叠氮桥化合物完全不稳定。通常,由于N 2消除的障碍要高得多,因此5,5'桥连的物质比其1,1'对应物更热稳定。除此之外,对于所有研究物种,都在W1-F12的理论水平上计算出了高精度的气相形成焓。
更新日期:2020-09-24
中文翻译:
带有长链氮链的Bis-Tetrazole和Bis-Triazole衍生物的热稳定性:高级量子化学计算的定量见解。
含有长链氮原子链的偶氮四唑和三唑衍生物作为有前途的绿色高能材料备受关注。但是,这些化合物通常表现出差的热稳定性和高冲击敏感性。初级分解反应的动力学和机理与这些问题直接相关。在当前的工作中,借助于高精度的CCSD(T)-F12量子化学计算,我们获得了许多N富集杂环的可靠键解离能和热解反应的激活势垒。我们研究了所有现有的含有N 10链的1,1'-偶氮双四唑,具有5,5'-桥接模式的对应物,以及带有azo-和乙氧基-桥的物种,它们通常是高能部分。N 8还考虑到含-偶氮双三唑。对于所有研究的化合物,发现自由基分解通道在动力学上是不利的。所有种类均通过开环反应分解,产生瞬态叠氮化物(或重氮)中间体,然后消除N 2。在偶氮双四唑衍生物的情况下,计算出的有效分解活化势垒为〜26–33 kcal mol –1,显着低于四唑(〜40 kcal mol –1)。这一事实与前一个物种的低热稳定性和高冲击敏感性非常吻合。N 2的激活障碍偶氮二化合物和母体四唑的消除几乎相同,有效的分解势垒由四唑-叠氮化物重排的热力学决定。与1,1'-偶氮双四唑相比,由于叠氮化物中间体缺乏π共轭,因此肼桥联的化合物在动力学上更稳定。继而,由于通过氢键形成的巨大的叠氮化物稳定性,叠氮桥化合物完全不稳定。通常,由于N 2消除的障碍要高得多,因此5,5'桥连的物质比其1,1'对应物更热稳定。除此之外,对于所有研究物种,都在W1-F12的理论水平上计算出了高精度的气相形成焓。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号