Nature Protocols ( IF 13.1 ) Pub Date : 2020-01-13 , DOI: 10.1038/s41596-019-0251-6 Máté E Maros 1, 2 , David Capper 3, 4 , David T W Jones 5, 6 , Volker Hovestadt 7, 8, 9 , Andreas von Deimling 3, 10 , Stefan M Pfister 5, 11, 12 , Axel Benner 13 , Manuela Zucknick 14 , Martin Sill 5, 11, 13
|
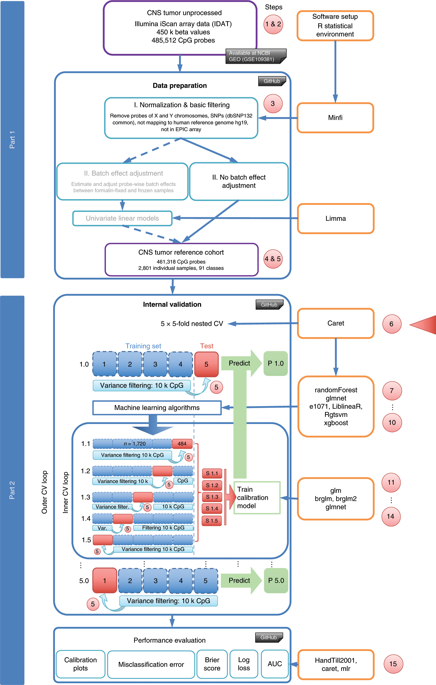
DNA methylation data-based precision cancer diagnostics is emerging as the state of the art for molecular tumor classification. Standards for choosing statistical methods with regard to well-calibrated probability estimates for these typically highly multiclass classification tasks are still lacking. To support this choice, we evaluated well-established machine learning (ML) classifiers including random forests (RFs), elastic net (ELNET), support vector machines (SVMs) and boosted trees in combination with post-processing algorithms and developed ML workflows that allow for unbiased class probability (CP) estimation. Calibrators included ridge-penalized multinomial logistic regression (MR) and Platt scaling by fitting logistic regression (LR) and Firth’s penalized LR. We compared these workflows on a recently published brain tumor 450k DNA methylation cohort of 2,801 samples with 91 diagnostic categories using a 5 × 5-fold nested cross-validation scheme and demonstrated their generalizability on external data from The Cancer Genome Atlas. ELNET was the top stand-alone classifier with the best calibration profiles. The best overall two-stage workflow was MR-calibrated SVM with linear kernels closely followed by ridge-calibrated tuned RF. For calibration, MR was the most effective regardless of the primary classifier. The protocols developed as a result of these comparisons provide valuable guidance on choosing ML workflows and their tuning to generate well-calibrated CP estimates for precision diagnostics using DNA methylation data. Computation times vary depending on the ML algorithm from <15 min to 5 d using multi-core desktop PCs. Detailed scripts in the open-source R language are freely available on GitHub, targeting users with intermediate experience in bioinformatics and statistics and using R with Bioconductor extensions.
中文翻译:
机器学习工作流程,用于估计 DNA 甲基化微阵列数据上精确癌症诊断的类别概率
基于 DNA 甲基化数据的精准癌症诊断正在成为分子肿瘤分类的最新技术。仍然缺乏为这些典型的高度多类分类任务选择关于经过良好校准的概率估计的统计方法的标准。为了支持这一选择,我们评估了成熟的机器学习 (ML) 分类器,包括随机森林 (RF)、弹性网络 (ELNET)、支持向量机 (SVM) 和增强树,并结合后处理算法并开发了 ML 工作流允许无偏类概率(CP)估计。校准器包括岭惩罚多项逻辑回归 (MR) 和通过拟合逻辑回归 (LR) 和 Firth 惩罚 LR 的 Platt 标度。我们使用 5 × 5 倍嵌套交叉验证方案在最近发表的 2,801 个样本和 91 个诊断类别的脑肿瘤 450k DNA 甲基化队列上比较了这些工作流程,并证明了它们对癌症基因组图谱的外部数据的普遍性。ELNET 是具有最佳校准配置文件的顶级独立分类器。最好的整体两阶段工作流程是 MR 校准的 SVM,其具有线性内核,紧随其后的是脊校准调谐 RF。对于校准,无论主要分类器如何,MR 都是最有效的。作为这些比较的结果而开发的协议为选择 ML 工作流程及其调整以生成校准良好的 CP 估计值以使用 DNA 甲基化数据进行精确诊断提供了有价值的指导。计算时间因 ML 算法而异,从 < 使用多核台式电脑需要 15 分钟到 5 天。开源 R 语言的详细脚本可在 GitHub 上免费获得,目标用户是在生物信息学和统计学方面具有中级经验并使用 R 和 Bioconductor 扩展的用户。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号