
本文以前文(https://zhuanlan.zhihu.com/p/149862369)为基础,对蛋白配体复合物分子模拟体系的结果进行一系列的粗浅分析,本文记述了简要的分析方法。
主体模拟结束之后,需要检查其温度、体系密度等体系参数符合预设要求。
通过 gmx energy命令提取模拟过程中随时间变化的体系参数数据,然后利用此命令给出的各参数平均值等数据,以及根据数据绘制得到的参数随时间变化的折线图,即可以判断体系是否在预设的参数条件下运行。
gmx energy -f md_results_1.edr -o output.xvg
# then choose the items that u wanna output同样还可以利用上述命令,提取体系potential、total-energy等参数信息进行分析。
轨迹文件中分子可能存在跨过周期性边界的情况,需要校正模拟体系的周期性。
首先将蛋白质和配体组合为一组,保存为 prolig_center.ndx,在其末尾添加如下内容:
[ center ]
500上述内容设定了周期性校正的中心原子,之后的校正会将此原子始终置于盒子中心,编号 500 的原子为蛋白质上某一个原子。
之后利用下述命令校正周期性:
gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -n prolig_center.ndx -o prolig_fit.xtc -pbc atom -center
# 如果需要消除热运动:
# gmx trjconv -f md.xtc -s md.tpr -fit rot+trans -o mdfit.xtc运行命令之后先选择校正中心为设置的center,然后选择对整个体系进行校正即可。
径向分布函数可以表征两种类型的分子之间的距离关系。分析配体到蛋白质表面的径向分布函数,可以获得稳定时期配体到蛋白质表面的距离等信息。
GROMACS中A类型粒子和B类型粒子之间的径向分布函数(RDF)被定义为:
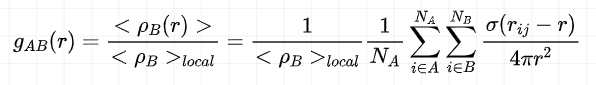
其中<\rho B(r)> 表示距离 A 粒子 r 距离,厚度为 bin 的壳层内B粒子的密度, <\rho B(r)> 表示以A粒子为中心,半径的球内B粒子的平均密度。
在本研究中 r_max 为4 nm,也即盒子边长的一半,bin 设置为 0.01 nm。
通常径向分布函数用于分析连续相(比如说模拟体系中的水分子在蛋白质周围的径向分布),但是也可以利用其对蛋白质周围配体的存在进行分析。根据上述公式可得,将其应用到配体分析时,函数会呈现出与连续相的径向分布函数不一样的特征。对于半径为 r 的球体,配体的平均密度是一个固定值,但在某些壳层内,因为没有配体分布,故而会使得函数值为零,而在某些配体密集分布的地方,会使得函数值较高;而对于连续相,则函数最终应该会收敛到1附近(也即在半径很大的时候,壳层平均密度与总体平均密度接近),且函数峰值不会太高。
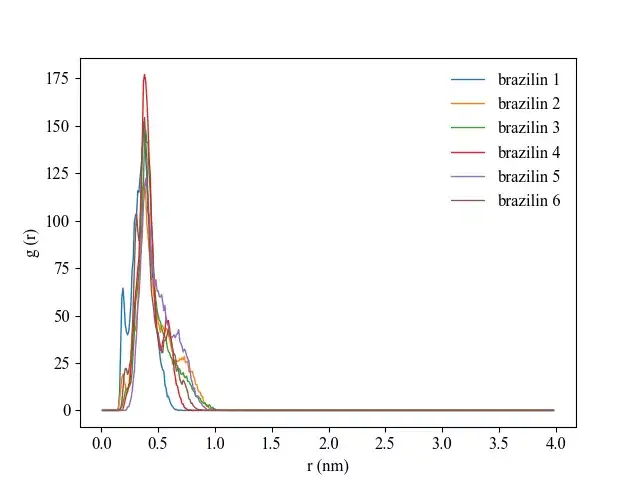
生成包含1ZIN的组ZIN.ndx,利用如下命令分析模拟稳定时期(模拟的最后15ns)配体1ZIN到蛋白质表面的径向分布函数:
gmx rdf -s prolig.tpr -f prolig_fit.xtc -o rdf_1_surf.xvg -n ZIN.ndx -ref protein -sel 1ZIN -bin 0.01 -surf mol -b 85000 -e 100000 -pbc yes结果图举例如下:
GROMACS将分子间的相互作用主要分为范德华相互作用和库伦相互作用;范德华力主要可以分为短程的Lenard-Jones potential(LJ)以及长程的色散相互作用;库伦相互作用则分为短程的库伦相互作用和长程的库伦相互作用。LJ以及库伦相互作用的短程和长程的截断值在上文的 md.mdp文件中都被设置为1.4 nm。范德华力随分子距离的增加衰减很快,所以短程的LJ势能占据了蛋白质和配体范德华作用的绝大部分,并且常被用作疏水相互作用的一种表征,本研究关于蛋白质和配体之间范德华力的研究就主要着眼于LJ势能。而库伦相互作用则一般认为是长程相互作用,且由于主体模拟中采用PME方法处理库伦作用,故而这里需要将短程库伦相互作用和倒易空间长程库伦相互作用加和才是最终的库伦相互作用。
在 gmx energy 给出的选项中,参数LJ(SR)表示短程LJ势能,Disper.corr.表示分子间作用力的色散项,Coulomb(SR)表示短程库伦相互作用,Coul.recip. 表示倒易空间的长程的库伦相互作用。本文抽提上述四种能量数据,另外计算总能量ETOTAL和库伦相互作用COULOMB,ETOTAL加和了上述四种能量,也即所有的范德华相互作用和库伦相互作用,而COULOMB则加和了Coulomb(SR)和Coul.recip,表示总的库伦相互作用。而被用于表征蛋白质和配体间的相互作用则主要是LJ(SR)、Coulomb(SR)以及ETOTAL。
前文的主体模拟设置中,我们取消了能量组的能量监测,这可以节约一定的算力,但是也导致我们最后得到的edr文件中并没有所需的蛋白质和配体相关的能量项,故而我们需要提取出蛋白质以及配体的轨迹并rerun得到蛋白质以及配体的相关能量项。
抽提蛋白配体能量项的步骤如下:
上述步骤抽提得到的是蛋白配体总体的能量,包含蛋白的能量、配体的能量以及蛋白配体之间的相互作用能,因而我们还需要利用相同的步骤抽提得到蛋白的相关能量以及配体的相关能量,然后利用蛋白配体的能量项对应减去蛋白的能量项和配体的能量项,差值极为蛋白和配体之间的相互作用能量。
本研究使用到的能量抽提脚本示例如下:
# shell scripts for energy extract
echo " ================ extract the prolig energy =================== "
gmx make_ndx -f npt.gro -o prolig.ndx
gmx trjconv -f md_results_1.xtc -s md_results_1.tpr -n prolig.ndx -o prolig.xtc
gmx convert-tpr -s md_results_1.tpr -n prolig.ndx -o prolig.tpr
gmx mdrun -s prolig.tpr -rerun prolig.xtc -e prolig.edr
gmx energy -f prolig.edr -o prolig_SR.xvg
echo " ================ extract the protein energy =================== "
gmx make_ndx -f npt.gro -o pro.ndx
gmx trjconv -f md_results_1.xtc -s md_results_1.tpr -n pro.ndx -o pro.xtc
gmx convert-tpr -s md_results_1.tpr -n pro.ndx -o pro.tpr
gmx mdrun -s pro.tpr -rerun pro.xtc -e pro.edr
gmx energy -f pro.edr -o pro_SR.xvg
echo " ================ extract the ligand energy =================== "
gmx make_ndx -f npt.gro -o lig.ndx
gmx trjconv -f md_results_1.xtc -s md_results_1.tpr -n lig.ndx -o lig.xtc
gmx convert-tpr -s md_results_1.tpr -n lig.ndx -o lig.tpr
gmx mdrun -s lig.tpr -rerun lig.xtc -e lig.edr
gmx energy -f lig.edr -o lig_SR.xvg利用python脚本进行能量的计算,得到最后的能量文件并作图,即可直观展示蛋白质和配体间的相互作用能随时间的变化。
以四种体系中配体与蛋白质之间的LJ(SR)为例:

均方根误差(RMSD)可理解为结构变化对于原子总数的平均;

RMSD 表示不同结构同一原子的距离。蛋白质的RMSD可以揭示蛋白质在模拟过程中的构象与初始构象之间的位置变化;蛋白质和配体的RMSD的变化趋势也是判断模拟是否达到稳定的重要表征。对于本研究而言,考虑到配体对于蛋白质可能的解聚作用,蛋白质的RMSD不仅可以表征蛋白质结构的稳定性,还可以进一步表征配体对于蛋白质是否具有一定的解聚作用。
之后提取RMSD数据:
gmx rms -s prolig.tpr -f prolig_fit.xtc -o rmsd.xvg运行命令之后选择抽提蛋白质的RMSD数据,以及蛋白质和配体的RMSD数据即可。
蛋白质的氨基酸一般包含很多柔性基团,故而抽提RMSD数据的时候可以选择碳链骨架,忽略氨基酸残基的影响。
还可以利用 rmsdist 程序分析每一帧原子之间距离变化的RMSD。
四种体系中蛋白质RMSD变化图举例:

RMSF为原子位置变化对于时间的平均,可以表征蛋白质氨基酸在整个模拟过程中的柔性和运动剧烈程度。
本研究可以借助RMSF探索蛋白质氨基酸的柔性,以及蛋白质柔性与配体结合活性氨基酸的关系。
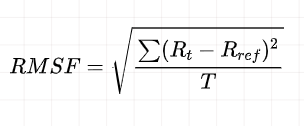
抽提RMSF的命令:
gmx rmsf -s prolig.tpr -f prolig_fit.xtc -o rmsd.xvg上述命令得到的数据是RMSF随原子序号的分布,不便于观察和分析。同样利用 rmsf 程序也可以做计算每个氨基酸的温度因子:
gmx rmsf -s prolig.tpr -f prolig_fit.xtc -o rmsf_temp.xvg -ox rmsf_avg.pdb -res -oq rmsf_bfac.pdb运行上述命令之后选择protein,之后通过pymol可视化rmsf_bfac.pdb ,通过pymol的b-factor来进行上色即可。
回旋半径可以表征蛋白质结构的紧实度,同样也可以依靠回旋半径来表征模拟过程中蛋白质的肽链松散程度的变化。计算整体回旋半径和单一坐标方向的公式如下:

使用校正过周期性的轨迹提取蛋白质的回旋半径数据的命令如下:
gmx gyrate -s prolig.tpr -f prolig_fit.xtc -o gyrate.xvg通过抽提蛋白质在模拟开始和结束时的三维结构并比较,可以表征蛋白质结构在模拟过程中的变化。
首先抽提固定帧(开始和结束的两帧并保存为两个pdb)
gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -o first.pdb -sep -b 0 -e 0 -pbc mol
gmx trjconv -s md_results_1.tpr -f md_results_1.xtc -o last.pdb -sep -b 100000 -e 100000 -pbc mol
# 选择蛋白质进行导出之后利用如下pymol脚本比较两个结构的差异并绘图。
load first0.pdb
load last0.pdb
load rmsf_hematoxylin.pdb
align last0, first0
align rmsf_hematoxylin, first0
run modevectors.py # modevectors.py 用于绘制豪猪图,可在pymol wiki下载
modevectors first0, last0
set bg_rgb, black
center
zoom
hide everything, first0
hide everything, last0
hide everything, rmsf_hematoxylin
show cartoon, rmsf_hematoxylin
spectrum b下图将RMSF也绘制在内,红色为氨基酸的RMSF,白色箭头从氨基酸在模拟零时刻时的位置指向氨基酸在模拟结束时的位置:

MD 体系中蛋白质和配体的运动轨迹可以直观地反映蛋白质和配体的运动以及结合情况,因而蛋白质和配体运动轨迹的可视化就尤为重要。
可以利用前述预平衡轨迹可视化的方法进行轨迹可视化,但是为了更方便地研究分子运动轨迹,将轨迹导出为pdb文件应该是更佳的方式。
使用如下命令从调整过周期性的模拟轨迹中导出pdb文件:
gmx trjconv -s prolig.tpr -f prolig_fit.xtc -o traj.pdb -dt 100
上述语句从已经校正过周期性边界的轨迹文件中每隔 100 ps 导出一帧到 pdb 文件,加入了蛋白质和配体的index文件可以只导出体系中的蛋白质和配体而忽略其它物质。
利用如下命令对载入pymol的pdb文件进行处理:
intra_fit prolig # 将轨迹中的帧对齐
dss state=1 # 计算二级结构并应用到所有帧
set all_states=1 # 叠合所有帧(optional)
viewport 640, 480 # 设置画框
set ray_trace_frames, 1 # 设置光线追踪之后将每一帧导出为图片并利用ffmpeg合并即可(当然能直接pymol导出为视频就更好了)
有的时候蛋白质的热运动有一些噪声,可以利用 filter 消除这些高频运动,只保留整体的运动。
gmx filter -s prolig.tpr -f prolig_fit.xtc -ol traj.pdb -dt 100 -n prolig.ndx -fit -nf 5将轨迹可视化之后,即可研究蛋白质和配体的相对运动,查看配体的结合位置、蛋白质的构象变化等等。
偶尔经过了周期性校正的轨迹导出的pdb文件在pymol无法正常cartoon显示,可以考虑换个周期性校正方式,比如说只保证分子完整,然后在pymol中对齐。
在模拟的过程中配体会与蛋白质形成一定数量的氢键,GROMACS提供了分子间氢键的分析工具,利用下述命令可以抽提出蛋白质和配体之间氢键数量随模拟时间的分布,lig.ndx为所有配体的组。
gmx hbond -s prolig.tpr -f prolig.xtc -n lig.ndx -num hbond_num.xvg -dt 100 -life hbond_life.xvg -ac hbond_ac.xvghbond程序使用几何准则来对氢键加以判定,当氢键供受体距离小于3.5埃且氢-供体-受体所成角度小于30度时,即认为其为一个氢键。
增加了 -ac 参数可以自动计算氢键的平均存在周期(forward lifetime),此参数可以作为衡量氢键稳定性的一个指标。
氢键分析这一块儿还有很多有待深入的地方,需要更多的学习和编程(或许将轨迹切成一帧帧的pdb然后利用PLIP分析也很不错)。
自由能形貌图(FEL,free energy landscape)可以表征物质在模拟过程中经历的自由能的变化。通过计算蛋白质和配体复合物的自由能形貌图可以给抽提复合物的特征构象提供指导。同时蛋白质在模拟的稳定时期的自由能形貌图可以表征蛋白质构象的稳定性。
FEL在GROMACS中利用两个变量来进行表示,通常是分子的回旋半径和均方根误差(文献中常见的做法应当是首先做「主成分分析」,然后利用前两个主成分作为FEL的变量),GROMACS可以利用这两个变量来计算相应的自由能;在得到回旋半径、均方根误差、自由能三个维度的数据之后,即可以作图得到自由能形貌图。GROMACS得到的自由能通常不是准确真实的自由能数据,而是基于构象的分布来进行的Gibbs自由能估计,因此充分的模拟采样是十分必要的。
构建自由能形貌图的过程如下:
gmx sham 命令获得自由能的xpm数据文件使用到的命令如下:
gmx gyrate -s prolig.tpr -f prolig_fit.xtc -o FEL_gyrate.xvg
gmx rms -s prolig.tpr -f prolig_fit.xtc -o FEL_rmsd.xvg
# 组合回旋半径和RMSD数据,生成 sham.xvg,使用 vim 的 visual block 模式,贼爽!
gmx sham -f sham.xvg -ls FEL_sham.xpm -b 8500 -tsham 310 -nlevels 100
# 利用jerkwin博客中的xpm2txt.py脚本将xpm转换为txt
python2 xpm2txt.py -f FEL_sham.xpm -o FEL_sham.txt
# 自己写个脚本把FEL_sham.txt作图就可以了如图(要是有时间渲染一下,少一些锯齿就更好了):

本文中涉及到一些自己写的 python、bash、pymol 脚本,以对分子动力学模拟和结果分析进行自动化以及一些简单的计算和绘图;有时间了这些代码将被重构和开源,预计不会耗时太久。可能重写为一个模块不是很合适,在考虑将之以零散的功能相对完善的脚本的形式开源。
继续加油~