
Structural biology incorporates techniques and principles of molecular biology, biochemistry and biophysics as a means of elucidating the molecular structure and dynamics of biologically relevant molecules. Recent progress in instrumentation has endorsed a new boost in structural biology as complex biological molecules can now be analyzed with unprecedented ease and efficiency. The three-dimensional structure of proteins and protein complexes provide great insights into the laws of life activities and mechanism of diseases, and thereby allowing rational design of novel diagnostic and therapeutic agents. There are three main research techniques for structural biology: single crystal X-ray diffraction (SC-XRD), nuclear magnetic resonance (NMR) and cryo-electron microscopy (Cryo-EM). However, there is no “all-purpose” method since all three of them offer unique advantages as well as limitations.
 Fig.1. Three main research techniques for structural biology. According to the statistics of PDB (https://www.rcsb.org/), more than 120,000 protein structures resolved by SC-XRD, accounting for nearly 90% of the total. And there are about 12,000 protein structures obtained by NMR. Although the total number of protein structures resolved by Cryo-EM is not comparable to that of the first two techniques, the explosive growth of structures from this technique is remarkable in recent years.
Fig.1. Three main research techniques for structural biology. According to the statistics of PDB (https://www.rcsb.org/), more than 120,000 protein structures resolved by SC-XRD, accounting for nearly 90% of the total. And there are about 12,000 protein structures obtained by NMR. Although the total number of protein structures resolved by Cryo-EM is not comparable to that of the first two techniques, the explosive growth of structures from this technique is remarkable in recent years.
1. Single crystal X-ray diffraction
X-ray crystallography uses X-ray to determine the position and arrangement of atoms in a crystal. The most classical method of X-ray crystallography is single crystal X-ray diffraction, in which crystal atoms cause the incident X-ray beam to produce scattered beams. When the scattered beams land on the detector, these beams produce a speckle diffraction pattern. As the crystal is gradually rotated, the angle and intensity of these diffracted beams can be measured, and then a three-dimensional image of the electron density within the crystal is generated. Based on this electron density, the average position of atoms in the crystal, chemical bonds, crystal barriers, and various information can be determined. For a single crystal with sufficient purity, homogeneity and regularity, the X-ray diffraction data can determine the average chemical bond angle and length to within a few tenths of a degree and to within a few thousandths of an angstrom, respectively.
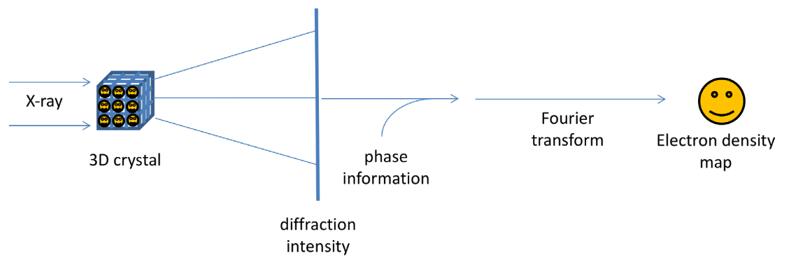 Fig.2. The physics and mathematical principles of X-ray crystallography to solve a structure
Fig.2. The physics and mathematical principles of X-ray crystallography to solve a structure
The single crystal X-ray diffraction technique was proposed and developed in 1912, and it has become the most important and useful tool for determining protein structure, since the protein structure of myoglobin was first determined in 1958. Nowadays, more than 140,000 protein structures have been deposited in protein databank (https://www.rcsb.org/), nearly 90% of which are resolved using single crystal X-ray diffraction technique, suggesting its advantages in studying the structure of biological macromolecule crystals.
The process of single crystal X-ray diffraction technique can be roughly divided into four steps. The first step is to obtain high-quality single crystals of the target protein, which is called protein crystallization. When the solution of the solubilized protein reaches supersaturation, it promotes protein aggregation and nucleation. Ultimately, individual protein molecules arrange themselves in a repeating series of unit cells by adopting a uniform orientation. Qualified crystals need to be of sufficient size (normally larger than 50 μm in all dimension) and quality (regular structure, no cracks or twins). Obtaining single crystals of high-quality is the limiting step to solve a structure with this method.
After obtaining a single crystal, a diffraction experiment is required. The crystal is immobilized in an intense X-ray beam, producing a diffraction pattern, which is recorded as the diffraction data (angle and intensity of the diffracted X-rays). As the crystal is gradually rotated, the previous reflections disappear and new reflections emerge. The diffraction intensity at each spot is recorded from each direction of the crystal.
Subsequently, the diffraction data obtained from the diffraction pattern are combined with various methods of structural analysis and data fitting to analyze the electron density distribution in the three-dimensional space within the unit cell.
In the last step, based on the electron density map, a model of atomic arrangement in the crystal can be produced and refined.
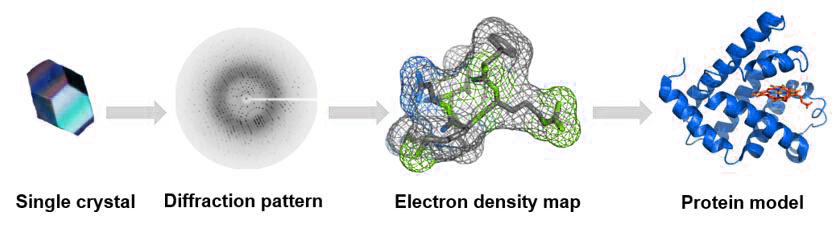 Fig.3. The process of single crystal X-ray diffraction technique
Fig.3. The process of single crystal X-ray diffraction technique
This method may yield high atomic resolution and is not limited by the molecular weight of the sample. It is suitable for water-soluble proteins, membrane proteins as well as macromolecular complexes. When manipulated properly, it becomes a powerful tool to deliver reliable structural data of biological macromolecules and determine the position and structure of the active center, and helps understand how the protein recognizes and binds ligand molecules at the atomic level.
However, the single crystal X-ray diffraction method also has several disadvantages. First, the sample must be crystallizable, but crystallization of biological macromolecules with large molecular weight can be difficult, particularly, membrane proteins are more challenging to crystallize because of its large size and poor solubilization. Second, an organized single crystal must be obtained to allow appropriate diffraction. Finally, the obtained three-dimensional structure of biological sample only represents a static form of the tested molecule (one of many possibilities), rather than a dynamic one.
2. Nuclear magnetic resonance (NMR)
The second method is nuclear magnetic resonance (NMR). Nuclei are charged, fast spinning particles, which are similar to outer electrons. The gyromagnetic ratios of different atomic nuclei are different and therefore have different resonance frequencies. The movement of the nucleus is not isolated--it interacts with the surrounding atoms both intra- and inter-molecularly. Therefore, through nuclear magnetic resonance spectroscopy, structural information of a given molecule can be obtained. Taking protein as an example, its secondary structure, such as α-helix, β-sheet, turn, circular, and curl, reflect the different arrangement of the main chain atoms of protein molecules three-dimensionally. The spacing of the atomic nuclei in different secondary domains, the interaction between nuclei, and the dynamic characteristics of polypeptide segments all directly reflect the three-dimensional structure of proteins. These nuclear features all contribute to spectroscopic behaviors of the analyzed sample, thus providing characteristic NMR signals. Interpretation of these signals by computer-aided methods leads to deciphering of the three-dimensional structure.
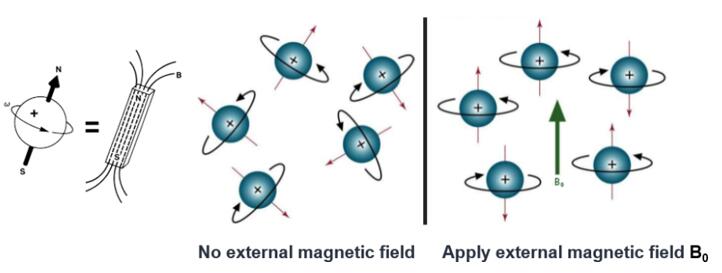 Fig.4. Nuclear spin
Fig.4. Nuclear spin
Since the first observation of condensed-state NMR signals in 1946, NMR technology has experienced a rapid development for over 70 years, and its application has been extended from the area of physics such as nuclear magnetic moment determination to chemistry, medicine, material science, life science and many others. Notably, in the 1980s, NMR technology was applied in the structural analysis of protein creatively, thus promoting the application of NMR in biological field. Although the amount of three-dimensional structure data of proteins obtained by NMR technology is not comparable to that of single crystal X-ray diffraction, the unique advantages of NMR technology have been widely noticed: NMR is able to provide information on a kinetic basis, such that the internal movement of proteins over multiple time scales and their binding mechanism to ligands can therefore be solved.
There are four main steps in an NMR experiment: sample preparation, data acquisition, spectral processing, and structural analysis. NMR analysis is performed on aqueous samples of protein with high purity, high stability, and high concentration. A sample volume ranging from 300 to 600 μL with a concentration range of 0.1-3 mM. The use of stable isotopes 15N, 13C and 2H for protein labeling can effectively increase signal intensity and resolution. Selective labeling of certain amino acids or chemical groups of proteins can greatly reduce signal overlap. Multidimensional NMR experiments are utilized to acquire information about the protein. The spectral processing is then performed to determine the atoms of the protein corresponding to each spectral peak on different NMR spectra. Finally, a series of spatially structured information such as NOE and J coupling constants are used to calculate the spatial structure using distance geometric or molecular dynamics methods.
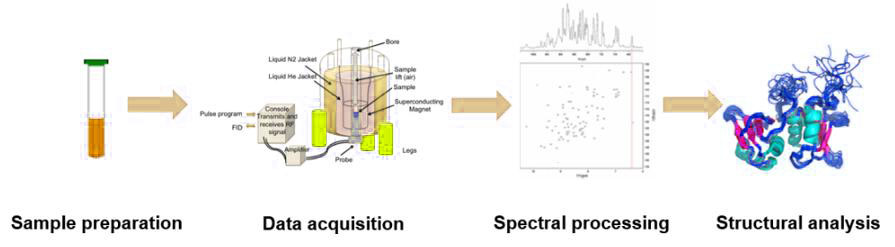 Fig.5. The process of nuclear magnetic resonance technology
Fig.5. The process of nuclear magnetic resonance technology
The most important feature of the NMR method is that the three-dimensional structure of macromolecules in the natural state can be measured directly in solution, and NMR may provide unique information about dynamics and intermolecular interactions. The resolution of the macromolecular three-dimensional structure can be as low as sub nanometer.
However, the NMR spectrum of biomolecules with large molecular weight is very complicated and difficult to interpret, thereby limiting the application of NMR in analyzing large biomolecules. Additionally, this technique requires relatively large amounts of pure samples (on the order of several mg) to achieve a reasonable signal to noise level.
3. Cryo-electron microscopy (Cryo-EM)
The third approach is the cryo-electron microscopy (Cryo-EM) technique, which includes three different methods: single particle analysis, electron tomography and electron crystallography. The essential mechanism of Cryo-EM is electron scattering. The basic principle is described as follows. Samples are prepared through cryopreservation prior to analysis. The coherent electrons are used as a light source to measure the sample. After the electron beam passes through the sample and the nearby ice layer, the lens system converts the scattered signal into a magnified image recorded on the detector. And signal processing is performed to obtain the three-dimensional structure of the sample.
Electron microscopy three-dimensional reconstruction technology was first discovered in 1968. The three-dimensional structure of T4 phage tail was reconstructed by electron micrographs. And then the general concept and methods of three-dimensional reconstruction of electron microscopy were proposed. For reducing the radiation damage, cryogenic electron microscopy was created in 1974. After more than 30 years of development, Cryo-EM has become a powerful tool for studying the structure of biological macromolecules. In recent years, the Cryo-EM technology has made revolutionary progress, particularly in the single particle analysis. Since 2013, with the tremendous advances in electron detector and image processing, Cryo-EM single particle analysis has progressed so rapidly that the resolution of Cryo-EM is now comparable to single crystal X-ray diffraction. At present, Cryo-EM is becoming a powerful tool for determining high resolution structure of biological macromolecules.
Before using the Cryo-EM to observe the sample, negative staining EM can be utilized for rapid screening of homogeneous sample. The Cryo-EM single particle analysis technique begins with the sample vitrification. During this process, the protein solution is instantly cooled, so that the water molecules do not crystallize, forming an amorphous solid. The frozen sample is then screened and data is collected in the system. A series of two-dimensional images can be taken during this period. Next, based on plenty of two-dimensional images acquired, particle alignment and classification are carried out. In the end, the data is processed by reconstruction software to generate a three-dimensional structural model.
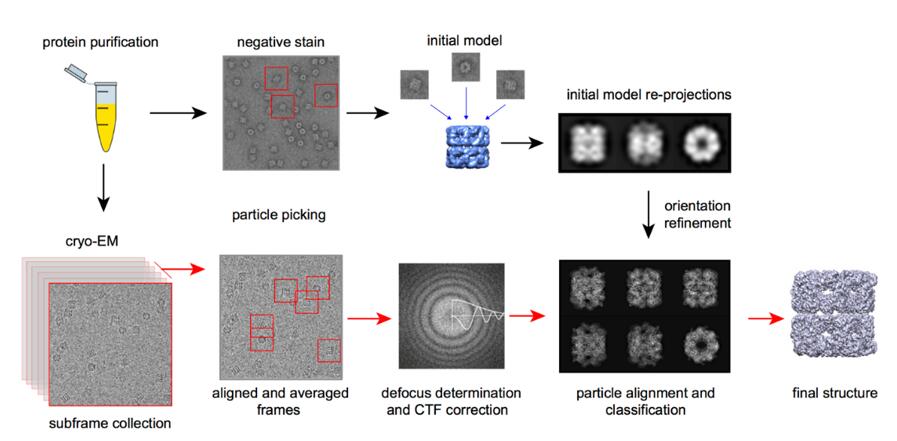 Fig.6. The process of Cryo-EM single particle analysis technique
Fig.6. The process of Cryo-EM single particle analysis technique
Compared to single crystal X-ray diffraction, the rapid freeze treatment of the sample maintains its closer-to-native state. Moreover, this method requires only a small amount of sample (about 0.1 mg), is more forgiven on sample purity, and does not need the protein to crystalize.
The main defect in this technique is that the particles are detected in unknown orientations. High levels of noise, due to the use of limited electron doses to minimize radiation damage, especially at high resolution, tends to complicate the determination of these orientations, and this is particularly a concern for smaller particles. Hence, structure determination of biological macromolecules by Cryo-EM was limited to large complexes or low-resolution models over the last few years.
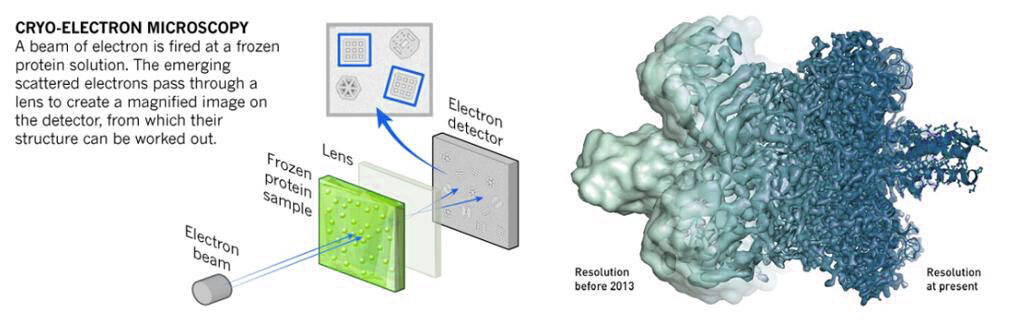 Fig.7. Cryo-electron microscopy
Fig.7. Cryo-electron microscopy
4. Summary
In summary, each technology has its own advantages in certain applications such that one method might be used extensively in some cases but rarely in others. Thus, understanding the nature of the analysis is the key in method selection. Not only will the inappropriate selection of method produce compromised results, it may also cause significant delays of the project, and result in financial losses. For more information, please see Table 1.
| Advantages | Disadvantages | Objects | Resolution | |
| X-ray Crystallography | • Well developed • High resolution • Broad molecular weight range • Easy for model building | • Difficult for crystallization • Difficult for diffraction • Solid structure preferred • Static crystalline state structure | • Crystallizable samples • Soluble proteins, membrane proteins, ribosomes, DNA/RNA and protein complexes | High |
| NMR | • High resolution • 3D structure in solution • Good for dynamic study | • Need for high sample purity • Difficult for sample preparation • Difficult for computational simulation | • MWs below 40–50 kDa • Water soluble samples | High |
| Cryo-EM | • Easy sample preparation • Structure in native state • Small sample size | • Relatively low resolution • Applicable to samples of high molecular weights only • Highly dependent on EM techniques • Costly EM equipment | • >150 kDa • Virions, membrane proteins, large proteins, ribosomes, complex compounds | Relatively Low (<3.5 Å) |
Table 1 The comparison of X-ray crystallography, NMR and Cryo-EM
References
from Internet
























Cyro-EM







