







Genes 2025, 16(4), 368; https://doi.org/10.3390/genes16040368 (registering DOI) - 23 Mar 2025
Abstract
Background: Broomcorn millet (Panicum miliaceum L.), a drought-tolerant C4 crop, is crucial for agricultural resilience in arid regions. Lipoxygenases (LOXs), key enzymes in plant stress responses, have not been studied in broomcorn millet. This study aimed to identify LOX genes in broomcorn
[...] Read more.
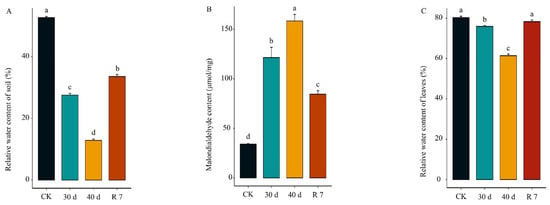
Background: Broomcorn millet (Panicum miliaceum L.), a drought-tolerant C4 crop, is crucial for agricultural resilience in arid regions. Lipoxygenases (LOXs), key enzymes in plant stress responses, have not been studied in broomcorn millet. This study aimed to identify LOX genes in broomcorn millet and elucidate their role in drought tolerance. Methods: We employed bioinformatics and physiological analyses to identify LOX genes in broomcorn millet. Expression profiles were assessed in different organs, and drought stress responses were evaluated in tolerant (HSZ, YXDHM) and sensitive (YS10) varieties. Antioxidant enzyme activities (SOD, POD, CAT) and malondialdehyde (MDA) levels were measured. Results: Twelve LOX genes were identified, classified into three subfamilies, and mapped across seven chromosomes. These genes contained stress-responsive cis-elements and showed organ-specific expression, with PmLOX5 exhibiting no detectable expression. Under drought stress, tolerant varieties showed elevated antioxidant activities and reduced MDA accumulation. PmLOX2, a homolog of Arabidopsis AtLOX1/AtLOX5, was significantly induced in tolerant varieties, correlating with enhanced antioxidant capacity and reduced oxidative damage. Conclusions:PmLOX genes, particularly PmLOX2, play a pivotal role in drought tolerance by modulating ROS scavenging and membrane protection. This study provides a foundation for leveraging LOX genes to improve drought resilience in broomcorn millet and related crops.
Full article
(This article belongs to the Section Bioinformatics)
►
Show Figures

Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}